
Dr. James Manos (MD)
January 5, 2016
Review: Tips in Medical Biochemistry
Volume (6)
CONTENTS
ELECTROLYTE DISTURBANCES (2)
Potassium (K+)
Hypokalemia
ECG (electrocardiographic) changes on hypokalemia
Diagnostic evaluation of hypokalemia (algorithm)
Hyperkalemia
Pseudohyperkalemia
ECG (electrocardiographic) changes on hyperkalemia
Diagnostic evaluation of hyperkalemia (algorithm)
Potassium urine; fractional excretion of potassium (FEK); trans-tubular potassium gradient (TTKG)
Fractional excretion of potassium (FEK) calculator
Transtubular potassium gradient (TTKG) calculator
Sodium (Na+)
Hyponatremia
Central pontine myelinolysis & rapid correction of hyponatremia
Diagnostic evaluation of hyponatremia (algorithm)
Syndrome of inappropriate antidiuretic hormone secretion (SIADH)
Fractional excretion of uric acid (FEUa) & SIADH
SIADH diagnostic algorithm
Hypernatremia
Diabetes insipidus (DI)
Polyuria & polydipsia algorithm
Diagnostic evaluation of hypernatremia (algorithm)
Sodium urine
Fluid balance
Fractional excretion of sodium (FENa)
Fractional excretion of sodium (FENa) calculator
Fractional excretion of urea (FEurea)
Fractional excretion of urea (FEurea) calculator
Phosphorus/ Phosphate (PO4–3)
Hypophosphatemia
Hypophosphatemia diagnostic algorithm
Hyperphosphatemia
Hyperphosphatemia diagnostic algorithm
Fractional excretion of phosphate (FEPi)
Fractional excretion of phosphate (FEPi) calculator
SERUM & URINE OSMOLALITY
Serum & Urine osmolality
Serum osmolality calculator
Urine osmolality calculator
Serum osmolal gap calculator
Urine osmolal gap calculator
Stool osmotic gap calculator
Potassium
· Potassium (K+): Potassium is the 8th or 9th most common element by mass (0.2%) in the human body; a 60 kg adult contains a total of about 120 g of potassium.
· Potassium cations are essential in neuron (brain & nerve) function; plays role in the influence of the resting cellular-membrane potential and the propagation of action potentials in neuronal, muscular, and cardiac tissue and in the regulation of the osmotic balance between cells & the interstitial fluid, with their distribution mediated in all animals (but not in all plants) by the Na+/K+ - ATPase (sodium-potassium adenosine triphosphatase) pump. This ion pump uses ATP to pump three sodium ions out of the cell and two potassium ions into the cell, thus creating an electrochemical gradient over the cell membrane.
· Also, the highly selective potassium ion (which are tetramers) are crucial for hyperpolarization, e.g., inside neurons, after an action potential is fired. A potassium intake sufficient to support life can, in general, be guaranteed by eating a variety of foods.
· Renal handling of potassium is closely connected to sodium handling. Potassium is the dominant cation (positive ion) inside animal cells [150 mmol/L, (4.8 g)], while sodium is the dominant cation of extracellular fluid [150 mmol/L, (3.345 g)]. In the kidneys, plasma is filtered through the glomeruli and into the renal tubules in enormous amounts (about 180 liters per day; so about 600 g of sodium and 33 g of potassium are filtered each day). All but the 1–10 g of sodium and the 1–4 g of potassium likely to be in the diet must be reabsorbed.
· Foods rich in potassium include potatoes, bananas, yam, parsley, dried apricots, dried milk, chocolate, various nuts (e.g., almonds), bamboo, shoots, avocados, coconut juice, soybeans, bran; it is also present in sufficient quantities in most fruits, vegetables, meat, and fish.
· Hypokalemia: it is generally defined as a serum potassium level of less than 3.5 mEq/L (3.5 mmol/L). Severe hypokalemia is a level of less than 2.5 mEq/L. Hypokalemia is a potentially life-threatening imbalance that may be iatrogenically induced.
· Signs & symptoms: patients are often asymptomatic, particularly those with mild hypokalemia. Symptoms that are present are usually from the underlying cause of the hypokalemia rather than the hypokalemia itself. The symptoms of hypokalemia are nonspecific and predominantly are related to muscular or cardiac function. Symptoms may include weakness and fatigue (most common); muscle cramps and pain (severe cases); worsening diabetes control or polyuria; palpitations; and psychological symptoms (e.g., psychosis, delirium, hallucinations, depression). Physical findings are often normal. Severe hypokalemia may manifest as bradycardia with cardiovascular collapse. Cardiac arrhythmias and acute respiratory failure from muscle paralysis are life-threatening complications that require an immediate diagnosis.
· Causes of hypokalemia:
· a) Inadequate potassium intake: eating disorders (anorexia, bulimia, starvation, pica, and alcoholism); dental problems (impaired ability to chew or swallow); poverty (inadequate quantity or quality of food eg, "tea-and-toast" diet of elderly individuals); and hospitalization (potassium-poor TPN (total parenteral nutrition)).
· b) Increased excretion of potassium, especially coupled with poor intake: It is the most common cause of hypokalemia.
· Increased potassium excretion may result from mineralocorticoid excess (endogenous or exogenous); hyperreninism from renal artery stenosis; osmotic diuresis by mannitol and hyperglycemia; increased gastrointestinal (GI) losses (vomiting, diarrhea, small intestine drainage, villous adenomas, VIPomas, and tropical illnesses such as malaria and leptospirosis); medications; genetic disorders.
· Endogenous sources of excess mineralocorticoid include Cushing syndrome; primary hyperaldosteronism (most commonly from an adrenal adenoma or bilateral adrenal hyperplasia); secondary hyperaldosteronism (volume depletion, congestive heart failure, cirrhosis, or vomiting); a tumor that is producing adrenocorticotropic hormone; genetic disorders.
· Exogenous causes of mineralocorticoid excess include steroid therapy for immunosuppression; glycyrrhizic acid (it inhibits 11-beta-hydroxysteroid dehydrogenase; contained in licorice and Chinese herbal preparations); type I & II renal tubular acidosis; hypomagnesemia.
· Drugs that can cause hypokalemia include: diuretics (carbonic anhydrase inhibitors, loop diuretics, thiazide diuretics; increased collecting duct permeability or increased gradient for potassium secretion can result in losses); methylxanthines [theophylline, aminophylline (used in asthma), caffeine], verapamil (with overdose; used for hypertension, cardiac arrhythmias & angina pectoris), quetiapine (an atypical antipsychotic; particularly in overdose); antibiotics (ampicillin, carbenicillin, high-dose penicillins; gentamicin); bicarbonate [used for acidosis, hyperkalemia & TCA (tricyclic antidepressants) overdose; may also be contained in antacids]; antifungal agents (amphotericin B, azoles, echinocandins), cisplatin (for cancer chemotherapy); ephedrine (from Ephedra; banned in the United States, but available over the internet; may be used as an appetite suppressant or decongestant); beta-agonist intoxication (beta – 1 such as dobutamine; used to treat cardiogenic shock & acute heart failure; beta – 2 agonists such as salbutamol (albuterol) are used for asthma).
· c) Genetic disorders: congenital adrenal hyperplasia (11-beta-hydroxylase or 17-alpha-hydroxylase deficiency); glucocorticoid-remediable hypertension; Bartter syndrome; Gitelman syndrome; Liddle syndrome; Gullner syndrome; glucocorticoid receptor deficiency; hypokalemic period paralysis; thyrotoxic periodic paralysis (TTPP); SeSAME syndrome (seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance).
· Bartter syndrome is a group of autosomal recessive disorders characterized by hypokalemic metabolic alkalosis, hypotension; and also sensorineural hearing loss. Mutations in 6 different renal tubular proteins in the loop of Henle have been discovered in individuals with clinical Bartter syndrome. Antenatal Bartter syndrome types 1, 2, 3, and 4A are inherited in an autosomal recessive manner. The most severe cases manifest antenatally or neonatally as profound volume depletion and hypokalemia. Patients with less severe cases present in childhood or early adulthood with persistent hypokalemic metabolic alkalosis that is resistant to replacement therapy. In general, however, the onset of true Bartter syndrome occurs by age 5 years. Patients with Bartter syndrome are more likely to have increased urine calcium excretion.
· Gitelman syndrome is an autosomal recessive disorder characterized by hypokalemic metabolic alkalosis and hypotension (low blood pressure). It is caused by a defect in the thiazide-sensitive sodium chloride transporter in the distal tubule, which is encoded by the SLC12A3 gene. Compared with Bartter syndrome, Gitelman's syndrome generally is milder and presents later. Moreover, Gitelman syndrome is complicated by hypomagnesemia & hypocalciuria (while patients with Bartter syndrome are more likely to have increased urine calcium excretion).
· Liddle syndrome is an autosomal recessive disorder characterized by a mutation affecting either the beta or gamma subunit of the epithelial sodium channel in the aldosterone-sensitive portion of the nephron. These subunits are encoded by theSCNN1G and SCNN1B genes and are inherited in an autosomal dominant fashion. Mutations to these genes lead to unregulated sodium reabsorption, hypokalemic metabolic alkalosis, and severe hypertension.
· Gullner syndrome is described as being like Bartter syndrome, except that renal histology showed normal juxtaglomerular apparatus and changes to the proximal tubules. Its identity is still unknown.
· Glucocorticoid receptor deficiency syndrome is caused by mutations to the theNR3C1 gene and has different clinical manifestations in patients who are homozygous than it does in those who are heterozygous. Homozygotes for this condition display mineralocorticoid excess, hypertension, hypokalemia, and metabolic alkalosis. Heterozygotes may have increased plasma cortisol levels and generally do not have hypokalemia or metabolic alkalosis. Reports have described likely heterozygotes for this condition who have symptoms of either partial adrenal insufficiency or mild virilization in females.
· Hypokalemic periodic paralysis types 1 and 2 are caused by mutations in the CACNL1A3 and SCN4A genes, respectively, and are both inherited in an autosomal dominant fashion. Patients with these disorders experience episodes of flaccid, generalized weakness, usually without myotonia. Patients will have hypokalemia during flaccid attacks.
· Thyrotoxic periodic paralysis (TTPP) is a form of hypokalemic periodic paralysis in which episodes of weakness associated with hypokalemia are seen in individuals with hyperthyroidism. It’s most common in Asian males.
· SeSAME syndrome (seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance). Hypokalemia, hypomagnesemia, hypocalciuria, and metabolic alkalosis are also seen. Moreover, some patients with SeSAME syndrome will have short stature, salt craving with polydipsia, renal potassium, and sodium wasting, and polyuria. This syndrome is inherited in an autosomal recessive fashion and is caused by mutations in the KCNJ10 gene, which encodes an inwardly rectifying potassium channel.
· d) Shift of potassium to the intracellular space: alkalosis (metabolic or respiratory); insulin administration or glucose administration (the latter stimulates insulin release); intensive beta-adrenergic stimulation; hypokalemic periodic paralysis; thyrotoxic periodic paralysis; refeeding (it is observed in prolonged starvation, eating disorders, and alcoholism), and hypothermia.
· Pseudohypokalemia: it is a decrease in the amount of potassium that occurs due to excessive uptake of potassium by metabolically active cells in a blood sample after it has been drawn. It is a laboratory artifact that may occur when blood samples remain in warm conditions for several hours before processing.
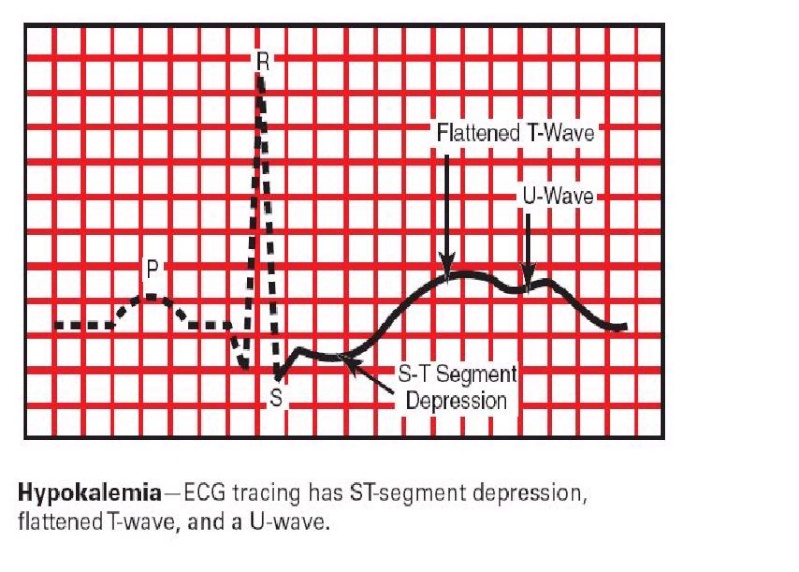
· ECG (electrocardiographic) changes on hypokalemia:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Diagnostic evaluation of hypokalemia (algorithm):
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Hyperkalemia: is defined as a serum potassium concentration greater than approximately 3.5 – 5.5 mEq/L in adults; the range in infants and children is age-dependent. Levels higher than 7 mEq/L can lead to significant hemodynamic and neurologic consequences, whereas levels exceeding 8.5 mEq/L can cause respiratory paralysis or cardiac arrest and can quickly be fatal.
· Signs & symptoms: many people with hyperkalemia are asymptomatic. When present, symptoms are non-specific and predominantly related to muscular or cardiac function. Weakness and fatigue are the most common complaints. Occasionally, patients may report the following: frank muscle paralysis, dyspnea (shortness of breath), palpitations, chest pain, nausea or vomiting, and paresthesias. In general, physical examination alone does not alerts the physician to the diagnosis of hyperkalemia, except when severe bradycardia is present, or muscle tenderness accompanies muscle weakness, suggesting rhabdomyolysis. Examination findings in patients with hyperkalemia include the following: vital signs usually normal, except occasionally in bradycardia due to heart block or tachypnea due to respiratory muscle weakness; muscle weakness and flaccid paralysis; depressed or absent deep tendon reflexes.
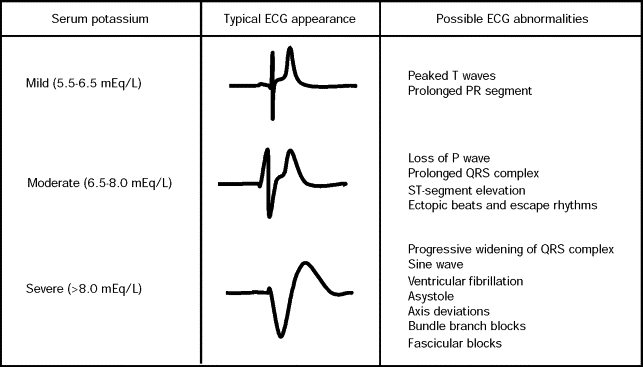
· ECG (electrocardiogram): Early ECG changes of hyperkalemia, typically seen at a serum potassium level of 5.5 – 6.5 mEq/L, includes: tall, peaked T waves with a narrow base, best seen in precordial leads; shortened QT intervals; ST-segment depression. At a serum potassium level of 6.5 – 8.0 mEq/L, the ECG typically shows: peaked T waves, prolonged PR interval, decreased or disappearing P wave, widening of the QRS, and amplified R wave. At a serum potassium level higher than 8.0 mEq/L, the ECG shows an absence of P wave, progressive QRS widening, and intraventricular/fascicular/bundle branch blocks. The progressively widened QRS eventually merges with the T wave, forming a sine wave pattern. Ventricular fibrillation (VF) or asystole (appearing as cardiac arrest) follows.
· Causes of hyperkalemia:
· a) Increased potassium intake. Alone, increased intake of potassium is a rare cause of hyperkalemia, because the mechanisms for renal excretion and intracellular disposition are very efficient. Generally, a relatively high potassium intake contributes to hyperkalemia in people who have impaired renal excretion or intracellular-to-extracellular shift.
· Causes of increased intake:
· i) High-potassium, low-sodium diets.
· ii) Ingestion of potassium supplements (the ingested amounts should have to be massive to be the sole cause of hyperkalemia, but even relatively small amounts can produce hyperkalemia in a patient with impaired renal excretion).
· iii) High concentrations of potassium in IV fluid preparations such as total parenteral nutrition formulas (where potassium is included).
· iv) Dietary salt substitutes (several “no-salt” or “low-salt” substitutes contain about 10-12 mEq of potassium per gram of salt and can be dangerous, especially with diminished renal function).
· v) Therapy with penicillin G potassium.
· vi) Packed red blood cells (PRBCs) transfusion (risk peaks at 2 – 3 weeks of cell storage).
· vii) Cardioplegia solutions (they contain 20 – 30 mmol/L of potassium chloride KCl).
· b) Decreased potassium excretion.
· i) Renal failure. Almost all patients who present with persistent hyperkalemia have impaired renal excretion of potassium. Mild degrees of renal failure generally do not result in resting hyperkalemia because of compensating mechanisms, however when the GFR (glomerular filtration rate) falls below 15 – 20 mL/min, significant hyperkalemia can occur, even in the absence of an abnormally large potassium load. The simple lack of nephron mass prevents normal potassium homeostasis (regulation).
· ii) Drugs.
· iii) Renal tubular acidosis.
· iv) Reduced distal sodium delivery.
· v) Reduced tubular fluid flow rate. Drug effects or renal tubular acidosis, can decrease renal potassium excretion and cause hyperkalemia even in individuals with normal or only mildly decreased renal function.
· Medications that can decrease potassium excretion include potassium-sparing diuretics (e.g. spironolactone, triamterene, amiloride), NSAIDs (nonsteroidal anti-inflammatory drugs), ACE (angiotensin-converting enzyme) inhibitors, angiotensin-receptor blockers (ARBs) (ACE inhibitors & ARBs are used for hypertension and for congestive heart failure (CHF)), cyclosporine & tacrolimus (immunosuppressants used e.g. in organ transplantation), pentamidine (an antibiotic against Pneumocystis jirovecii that causes pneumonia on patients with AIDS & also used against trypanosomiasis), trimethoprim-sulfamethoxazole (an antibiotic), heparin (a blood thinner), ketoconazole (an antifungal), metyrapone (a drug used in the diagnosis of adrenal insufficiency and occasionally in the treatment of Cushing’s syndrome), and some herbs.
· Disorders that can cause type IV renal tubular acidosis include diabetes mellitus, sickle cell disease or trait, lower urinary tract obstruction, adrenal insufficiency, primary Addison syndrome (autoimmune or due to tuberculosis or infarct), enzyme deficiencies, and genetic disorders.
· c) Shift of potassium into extracellular space: this is rarely the sole cause of hyperkalemia, because the mechanisms for renal excretion are very efficient, except in people who have impaired renal excretion.
· Factors that can shift potassium into the extracellular space include metabolic acidosis, beta-adrenergic blockade, acute tubular necrosis, electrical & thermal burns, cell depolarization, head trauma, digitalis toxicity, fluoride toxicity, cyclosporine (immunosuppressant used e.g. in organ transplantation), methotrexate (an antimetabolite & antifolate; used for cancer, autoimmune diseases, ectopic pregnancy and for induction of medical abortions), propofol (an anesthetic) infusion syndrome, rhabdomyolysis, tumor lysis syndrome, and succinylcholine use (also known as suxamethonium; used as a muscle relaxant and for short-term paralysis on endotracheal intubation). Hypertonicity may lead to hyperkalemia by the loss of intracellular water, resulting in an increased intracellular potassium concentration, favoring a gradient for potassium to move out of the cells; also as water exits the cells, sweeps potassium along.
· Causes of hyperosmolality include hyperglycemia in uncontrolled diabetes mellitus (the most common cause); hypernatremia, and hypertonic mannitol. There is also some evidence that long-term aldosterone deficiency impairs cell potassium uptake. Toad venom, which is used in traditional Chinese medicine and in folk medicine in southeastern Asia, contains cardiac glycosides whose structure and biochemical activity are similar to those of digitalis. These can cause hyperkalemia. Toad venom is prepared from dried secretions from the Asiatic toad (Bufo gargarizans). It is also an ingredient in Chinese medications (e.g., Chan Su, Lu-Shen Wan), toad venom has also turned up in purported aphrodisiacs.
· d) Diabetes mellitus (DM). Patients with diabetes constitute a high-risk group for hyperkalemia: they develop defects in all aspects of potassium metabolism. The typical healthy diabetic diet often is high in potassium and low in sodium. Diabetic persons frequently have underlying renal disease and often develop hyporeninemic hypoaldosteronism (i.e., decreased aldosterone secondary to suppressed renin levels), impairing renal excretion of potassium. Many patients with diabetes are placed on ACE inhibitors (angiotensin-converting enzyme inhibitors) or ARBs (angiotensin-receptor blockers) therapy for the treatment of hypertension or diabetic nephropathy, exacerbating the defect in potassium excretion. Finally, persons with diabetes have insulin deficiency or resistance to insulin action, limiting their ability to shift potassium intracellularly.
· e) Genetic disorders. Genetic disorders that can result in hyperkalemia include:
· i) Glomerulopathy with fibronectin deposits (GFND). GFND is a genetically heterogeneous autosomal dominant disorder which manifests as proteinuria, hypertension & type IV renal tubular acidosis. It eventually leads to end-stage renal failure, in the second to fifth decade of life. Type 1 GFND is related to chromosome 1q32. However, the gene is unknown at this time. Type 2 GFND is caused by mutations in the FN1 gene located on chromosome 2q34.
· ii) Disorders of steroid metabolism and mineralocorticoid receptors. 21-hydroxylase deficiency in its classic form and aldosterone synthase deficiency result in hyperkalemia due to low aldosterone levels. 11-Beta hydroxylase deficiency, 3-beta-hydroxysteroid dehydrogenase deficiency, and 17 alpha-hydroxylase/17,20-lyase deficiency are generally not characterized by the development of hyperkalemia.
· iii) Congenital hypoaldosteronism. It is caused by mutations in the CYP11B2 gene, which encodes the type II corticosterone methyl oxidase enzyme. It is inherited in an autosomal recessive manner. Patients with this disorder have decreased aldosterone and salt wasting. They will have an increased serum ratio of 18-hydroxy corticosterone to aldosterone.
· iv) Pseudohypoaldosteronism.
· Type I pseudohypoaldosteronism (PHAI) can be caused by an inactivating mutation of 1 of 3 encoding subunits of the epithelial sodium channel (SCNN1A, SCNN1G, or SCNN1B).
· PHAI is inherited in an autosomal recessive manner. These mutations result in impaired potassium secretion due to impaired sodium reabsorption in the distal tubule. PHAI tends to be most severe in the neonatal period, causing renal salt wasting and respiratory tract infections. Sweat, stool, and saliva have high sodium concentrations. Sometimes this disorder can be mistaken for cystic fibrosis. Another form of PHAI is caused by mutations in the NR3C2 gene and is inherited in an autosomal dominant manner. Patients with this disorder may present in the neonatal period with renal salt wasting and hyperkalemic acidosis similar to those seen in the autosomal recessive form. Patients with this form of PHAI generally improve with age and are typically asymptomatic in adulthood.
· Gordon syndrome, or pseudohypoaldosteronism type II (PHAII), characterized by hyperkalemia and hypertension, is caused by mutations in several genes. The genes causing this disorder code for protein kinases that are localized to the distal tubule and that regulate ion transport in this nephron segment. WNK4 appears to have several roles in regulating sodium, potassium, and chloride transport through transcellular and paracellular pathways. PHAII from mutations in WNK1 is significantly less severe than PHAII from mutations in WNK4 or KLHL3, whereas PHAII from mutations in CUL3 is more severe.
· All forms of PHII generally respond to treatment with thiazide diuretics.
· v) Disorders of chloride homeostasis. Isolated hyperchlorhidrosis is caused by mutations in the CA12 gene, and is inherited in an autosomal recessive manner. This disorder can cause excessive salt wasting in sweat, which can result in severe hyponatremic dehydration and hyperkalemia.
· vi) Nephronophthisis. It is characterized by enlargement of the kidneys, inflammatory portal fibrosis of the liver, and variable development of end-stage renal disease (ESRD). Patients with the infantile form of this disease generally reach ESRD before the age of 2 years. Patients with the juvenile form reach ESRD at a median age of 13 years. Patients with other forms of the disease have a variable natural history. Eventually, this disorder causes progressive interstitial fibrosis and tubulopathy. Routine laboratory evaluation will show increased creatinine and potassium.
· vii) Hyperkalemic periodic paralysis (HYPP). HYPP is caused by mutations in the SCN4A gene and is inherited in an autosomal dominant manner. During attacks (which can be precipitated by the administration of potassium), individuals with HYPP have flaccid generalized weakness and increased serum potassium levels. Patients with HYPP can also have myotonia, which is not typically a feature of hypokalemic periodic paralysis (HOKPP). Some similar disorders involving myotonia or muscular weakness are allelic to HYPP.
· Pseudohyperkalemia: Pseudohyperkalemia occurs when laboratory reports of potassium do not reflect actual values. The most common cause is hemolysis (lysis of red cells) in a phlebotomy specimen. Pseudohyperkalemia is a rise in serum potassium concentration with concurrently normal plasma potassium concentration. It is an in vitro phenomenon. Lately, pseudohyperkalemia was defined when serum potassium concentration exceeded that of plasma by more than 0.4 mmol/L provided that samples are collected under strict techniques, remain at room temperature, and are tested within 1 hour from blood specimen collection.
· Hemolysis of red blood cells releases large amounts of potassium into the surrounding plasma. Erythrocytes contain 23 times as much potassium as the plasma.
· The most common causes of hemolysis are related to mechanical factors during the collection process:
· Use of a syringe with excessive suction applied to the plunger is by far the most common cause of hemolysis, with almost 80% of hemolyzed samples associated with the use of a syringe rather than an evacuated tube for collection. In one study, 19% of syringe collected specimens were hemolyzed, as compared to 3% of specimens that were collected in evacuated tubes.
· Forcibly squirting the blood from a syringe into an evacuated tube causes shear forces on the red cell membrane, resulting in rupture of the cell. Evacuated tubes should be allowed to fill slowly from the vacuum in the tube, without pressing on the syringe plunger.
· Drawing the blood through a small needle or catheter also ruptures red cells as they pass through either. The narrower the needle or catheter, the greater is the hemolysis. Blood collected with a 23-gauge needle has higher potassium concentrations than blood from the same individual collected with a 19-gauge needle. The hemolysis rate is inversely proportional to the diameter of the needle or catheter, with the highest hemolysis rates in 24- to 20-gauge catheters. Using a large-diameter needle that causes the blood to enter the evacuated tube with great force also can rupture red cells. Becton-Dickinson recommends using a special low-vacuum evacuated tube in this situation.
· Drawing the blood through an IV tube or catheter where the diameters of the catheter, tube adapter device, and cap-piercing needle are mismatched can cause turbulence of the blood, with cell rupture.
· Inverting the tube too vigorously to mix the blood with an anticoagulant also causes turbulence.
· Some authors have listed the prolonged application of a tourniquet as a cause of hemolysis, or elevation of the potassium without hemolysis.
· Because the major cause for hemolysis is the use of a syringe rather than an evacuated tube system for blood drawing, it is mostly a problem in areas where blood is drawn by non-laboratory personnel, such as in emergency departments and ICUs.
· Several studies have shown that hemolyzed serum or plasma containing 1 gram/Liter of hemoglobin will have an increase of 0.27 mmol/L to 0.33 mmol/L of potassium.
· Other cellular constituents, such as lactate dehydrogenase, ALT (sGPT), AST (sGOT), and CK (CPK) are also increased.
· Other causes of pseudohyperkalemia include:
· a) Erythrocytosis [the increase in total red blood cell mass secondary to any of a number of non-hematological systemic disorders in response to a known stimulus (secondary polycythemia, may be seen in persons living at high altitudes, and also patients with cardiopulmonary problems, e.g., heart failure, congenital heart disease, pulmonary emphysema and respiratory insufficiency) in contrast to primary insufficiency (polycythemia vera), a myeloproliferative disease]
· b) Leukocytosis (a white blood cell (leukocyte) count above the normal range in the blood; it is frequently a sign of an inflammatory response, most commonly the result of infection, but may also occur on a blood malignancy)
· c) Thrombocytosis [the presence of high platelet counts in the blood, and can be either primary (also termed essential and caused by a myeloproliferative disorder) or reactive (also termed secondary)], or mixed disorders. Potassium released from platelets can lead to spuriously high levels of potassium in a blood sample allowed to clot to collect serum.
· Pseudohyperkalemia can be excluded by repeating the sample collection as atraumatically as possible and obtaining serum and plasma potassium levels. In patients with pseudohyperkalemia, the plasma potassium will be normal in the face of elevated serum potassium.
· Pseudohyperkalemia also is positively correlated to (1) thrombocytosis due to the release of potassium from platelet granules during coagulation, (2) erythrocytosis due to the dilution of the released potassium in smaller volumes of serum, and (3) the presence of activated platelets, which have the capability of aggregation at a higher speed and release more potassium during degranulation.
· However, pseudohyperkalemia may be "masked" when in a state of hypokalemia because potassium moves back into the intracellular space in vitro, and the phenomenon is ameliorated or even not detected.
· A human study concluded that pseudohyperkalemia is mainly present in patients with thrombocytosis or mixed-type disorders, probably as a result of the degranulation of platelets, which offers a potassium load to the surrounding plasma at the time of clot formation in vitro. However, the degree of pseudohyperkalemia does not increase proportionally with the increase of platelet counts, which may be associated with the transfer of part of potassium load from the plasma back into red and white blood cells.
· Another human study concluded that the difference between serum and plasma potassium concentration (Dk) is increased in patients with erythrocytoses, thrombocytoses, or both. This phenomenon is more profound in patients with a mixed type disorder, such as polycythemia vera patients, compared to those with erythrocytoses alone.
· Other causes of pseudohyperkalaemia:
· Using plasma reference ranges to interpret serum values could result in pseudohyperkalemia.
· Factitious hyperkalemia may be seen in WBC neoplasms due to increased membrane fragility and little reserve capacity for withstanding mechanical agitation or by leakage into the serum. It is suspected that neoplastic WBC membranes (e.g., chronic lymphocytic leukemia) are more likely to be leaky or to be disrupted during pneumatic chute transport. Also, at high levels of leukocytosis, there is increased consumption (and thereby exhaustion) of metabolites that fuel the ATP pump.
· Reverse pseudohyperkalemia is when plasma potassium is falsely high, but the serum potassium is normal. This phenomenon has been reported in samples of patients with leukemia/lymphoma. In one study, the plasma potassium concentration of a sample collected in a lithium heparin tube was 6.0 mmol/L higher than simultaneously measured serum potassium concentration. The degree of increase in potassium was directly related to the amount of heparin.
· As the spleen is a significant reserve for platelets, post-splenectomy status has also been reported to be associated with pseudohyperkalemia.
· Potassium containing IV fluids are common contaminants as are potassium salts of tube additives. If the recommended order of draw during phlebotomy is not maintained, carryover and backflow of potassium salts of tube additives such as EDTA or oxalate can elevate measured potassium.
· If ethanol containing antiseptics are not allowed to dry completely before venipuncture, the solution can enter the bloodstream and disrupt cell membranes.
· Povidone-iodine (e.g., Betadine (R)) in one study has been associated with an increase in measured potassium up to 1 mmol/L. However, the mechanism is unclear.
· Temperature: the recommended temperature for specimen storage before testing is 15 – 25°C. Specimens should not be stored between 2°C and 8°C, or above room temperature for more than 24 h.
· Time: delayed processing, results in exhaustion of available glucose to generate ATP. Since ATP fuels the sodium-potassium pump and maintains the gradient across the cell membrane, failure of the pump results in leakage of potassium out of the cell, resulting in pseudohyperkalemia.
· Fear of imminent venipuncture or crying associated with hyperventilation (even for 3 – 6 min) is associated with acute respiratory alkalosis, which results in a significant hyperkalemic response mediated by enhanced alpha-adrenergic activity
· Reference: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3662091/
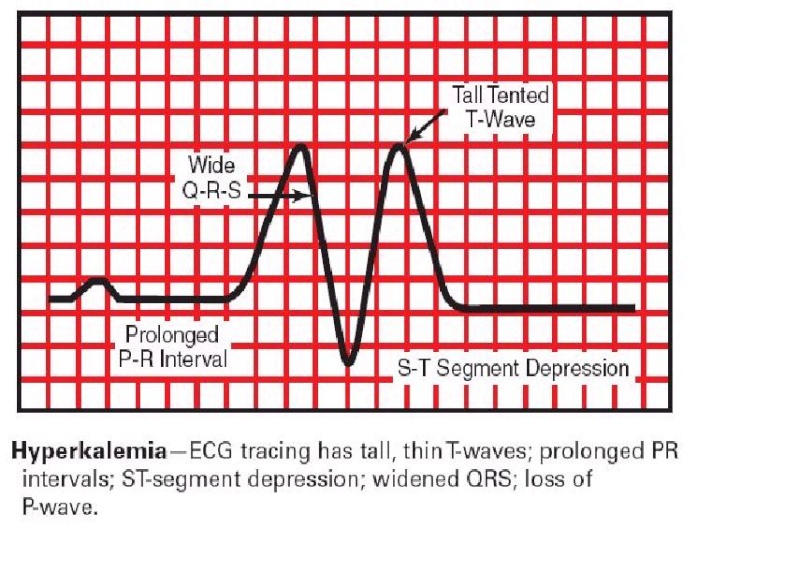
· ECG (electrocardiographic) changes on hyperkalemia
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Diagnostic evaluation of hyperkalemia (algorithm)
{kind=link}
{kind=link}
{kind=link}
· Potassium urine; fractional excretion of potassium (FEK); trans-tubular potassium gradient (TTKG): in healthy people nearly all potassium filtered by the kidney is reabsorbed. Potassium excretion reflects distal tubule secretion of potassium, which is stimulated by aldosterone, and the rate of potassium entry into the plasma from the diet and from cells.
· Urine potassium levels are generally helpful only in the evaluation of patients with unexplained hypokalemia. The reference range is 25 – 123 mEq/24 hours. The most accurate measurement of urine potassium is obtained with a 24-hour urine collection, collected in a container without preservative. Specimen should be refrigerated during and after the collection.
· Interpretation of urine potassium levels: urine potassium levels between 0 –10 mEq/L suggest the GI (gastrointestinal) tract is the source of potassium loss, while levels >10 mEq/L suggest renal potassium loss.
· An alternative method to assess urine potassium excretion is to collect a random urine sample and calculate the fractional excretion of potassium (FEK). FEK reflects the relationship of potassium excreted to the amount that has been filtered by the kidneys and requires measurement of potassium and creatinine in both urine and plasma.
· The formula is: FEK= (urine potassium x plasma creatinine x 100)/plasma potassium x urine creatinine
· Interpretation of fractional excretion of potassium (FEK):
· a) In patients with hyperkalemia, a FEK < 10% suggests that hyperkalemia is due to renal (kidney) disease. A value > 10% indicates that hyperkalemia is due to extrarenal causes.
· b) In patients with hypokalemia, a FEK <10% suggests that hypokalemia is due to extrarenal causes, while a FEK value > 10% indicates that the underlying cause is of renal origin.
· Urine potassium loss can also be evaluated by calculating the trans-tubular potassium gradient (TTKG) using the following formula:
TTKG = urine potassium/(plasma osmolality/urine osmolality)/serum potassium
TTKG = urine potassium/(plasma osmolality/urine osmolality)/serum potassium
· Note: this formula is accurate when urine osmolality is higher than plasma osmolality, and urine sodium is greater than 25 mEq/L.
· Interpretation of TTKG:
· a) Individuals with hyperkalemia or high potassium intake and normal renal function should excrete potassium into the urine resulting in a TTKG > 10. Values < 7 are consistent with mineralocorticoid deficiency, especially if accompanied by hyponatremia and high urine sodium concentration.
· b) Individuals with hypokalemia should have TTKG values < 2. Higher values are consistent with inappropriate stimulation of potassium secretion.
· Fractional excretion of potassium (FEK) calculator:
· Transtubular potassium gradient (TTKG) calculator:
· Sodium (Na+ ): is a chemical element with the symbol Na (Latin: natrium) and atomic number 11. It is a soft, silver-white, highly reactive metal and is a member of the alkali metals. Its only stable isotope is 23Na. In humans, sodium is an essential nutrient that regulates blood volume, blood pressure, osmotic equilibrium, and PH. The minimum physiological requirement for sodium is 500 milligrams per day.
· Sodium chloride (NaCl) is the principal source of sodium in the diet, and is used as seasoning and preservative; most of it comes from processed foods.
· The tolerable upper intake levels (UL) for sodium is 2.3 grams per day. This threshold could lead to hypertension when exceeded (however, on average people in the USA consume 3.4 grams per day).
· Salt contains about 39.3% sodium (the rest being chlorine and other trace chemicals); thus the UL of 2.3g sodium would be about 5.9g, or 2.7ml of salt—about half a US teaspoon (1/3 tablespoon; 5 mL).
· The renin-angiotensin system regulates the amount of fluids and sodium in the body. Reduction of blood pressure and sodium concentration in the kidney result in the production of renin, which in turn produces aldosterone and angiotensin, retaining sodium in the urine. Because of the increase in sodium concentration, the production of renin decreases, and the sodium concentration returns to normal.
· Sodium is also necessary for neuron function and osmoregulation between cells and the extracellular fluid; their distribution is mediated in all animals by Na+ /K+-ATPase (sodium-potassium adenosine triphosphatase, also known as a Na+/K+ pump (sodium-potassium pump)); hence, sodium is the most prominent cation in extracellular fluid.
· Blood sodium testing is used to detect abnormal concentrations of sodium, termed hyponatremia (low sodium) and hypernatremia (high sodium).
· Urine sodium levels are typically tested in patients who have abnormal blood sodium levels to help determine whether an imbalance is from, for example, taking in too much sodium or losing too much sodium. Urine sodium testing is also used to see if a person with hypertension is overeating salt. It is often used in persons with abnormal kidney tests to help the doctor determine the cause of kidney damage, which can help guide treatment.
· Hyponatremia: patients may present to medical attention with symptoms related to low serum sodium concentrations. However, many patients present due to manifestations of other medical comorbidities, with hyponatremia being recognized only secondarily. Thus, for many people, the recognition of hyponatremia is entirely incidental. Many medical illnesses, such as congestive heart failure, liver failure, renal failure, or pneumonia, may be associated with hyponatremia. These patients frequently present because of primary disease symptomatology.
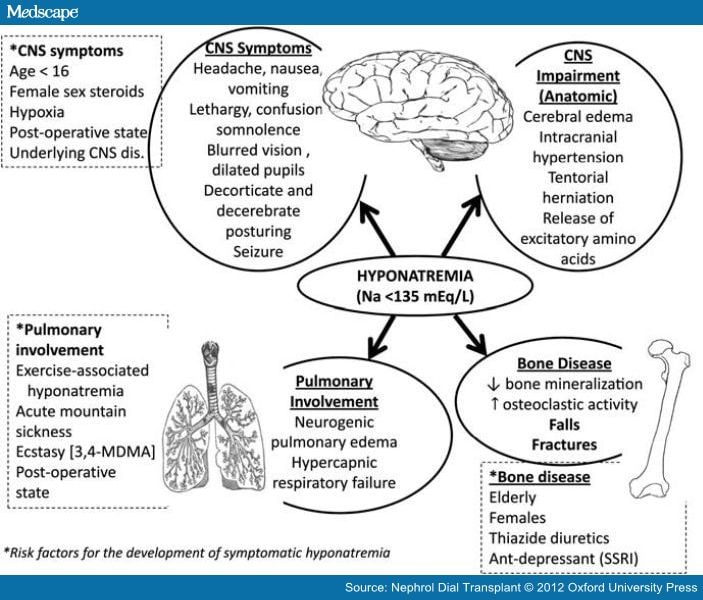
· Signs & symptoms: Symptoms of hyponatremia range from nausea and malaise, with mild reduction in the serum sodium, to lethargy, decreased level of consciousness, headache, and (if severe) seizures and coma. Overt neurologic symptoms most often are due to deficient serum sodium levels (usually < 115 mEq/L), resulting in intracerebral osmotic fluid shifts and brain edema. This neurologic symptom complex can lead to tentorial herniation with subsequent brain stem compression and respiratory arrest, resulting in death in the most severe cases. The severity of neurologic symptoms correlates well with the rate and degree of the drop in serum sodium. A gradual drop in serum sodium, even to very low levels, may be tolerated well if it occurs over several days or weeks, because of neuronal adaptation. The presence of underlying neurologic disease, like a seizure disorder, or non-neurologic metabolic abnormalities, like hypoxia, hypercapnia, or acidosis, also affects the severity of neurologic symptoms.
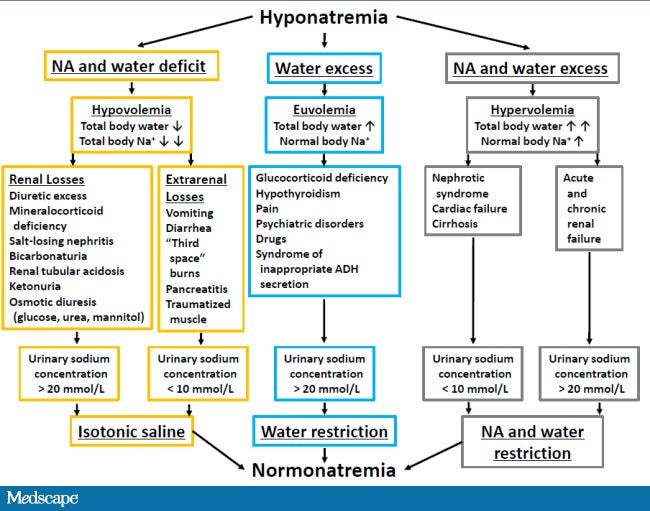
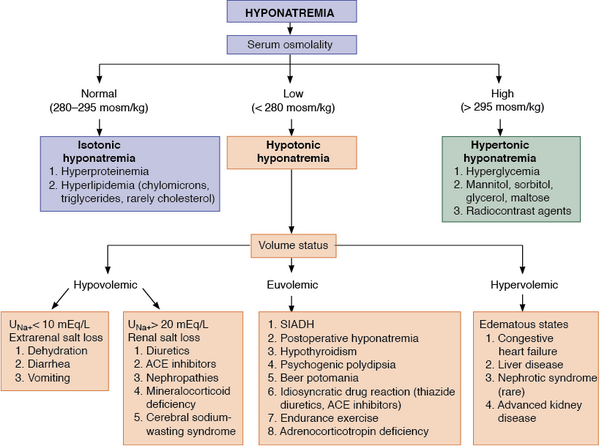
· Causes of hyponatremia:
· a) Hypertonic hyponatremia. Patients with hypertonic hyponatremia have normal total body sodium and a dilutional drop in the measured serum sodium due to the presence of osmotically active molecules in the serum, which causes a water shift from the intracellular compartment to the extracellular compartment. Glucose produces a drop in the serum sodium level of 1.6 mEq/L for each 100 mg/dL of serum glucose greater than 100 mg/dL. This relationship is non-linear, with a greater reduction in plasma sodium concentrations with glucose concentrations over 400 mg/dL, making 2.4 mEq/L for each 100 mg/dL increase in glucose over 100 mg/dL a more accurate correction factor when the glucose is greater than 400 mg/dL. Other examples of osmotically active molecules include mannitol (often used to treat brain edema) or maltose (used with intravenous immunoglobulin administration).
· b) Normotonic hyponatremia. Severe hyperlipidemia and paraproteinemia can lead to low measured serum sodium concentrations with normal serum osmolality. Normally, the plasma water comprises 92 – 94% of plasma volume. The plasma water fraction falls with an increase in fats and proteins. The measured sodium concentration in the total plasma volume is respectively reduced, although the plasma water sodium concentration and plasma osmolality are unchanged. This artifactual low sodium (so-called pseudo-hyponatremia) is secondary to measurement by flame photometry. It can be avoided by direction-selective electrode measurement. Hyponatremia post-transurethral resection of the prostate (TURP) or hysteroscopy is caused by absorption of irritants, glycine, sorbitol, or mannitol, contained in non-conductive flushing solutions used. The degree of hyponatremia is related to the quantity and rate of fluid absorbed. The plasma osmolality is also variable and changes over time. The presence of a relatively large osmolar gap due to excess organic solute is diagnostic.
· c) Hypotonic hyponatremia. Hypotonic hyponatremia always reflects the inability of the kidneys to handle the excretion of free water to match intake. It can be divided pathophysiologically according to the effective intravascular volume into hypovolemic, hypervolemic, and euvolemic.
· I) Hypovolemic hypotonic hyponatremia: it usually indicates concomitant solute depletion, with patients presenting with orthostatic symptoms. In the setting of decreased intravascular volume (e.g., severe hemorrhage or severe volume depletion secondary to gastrointestinal or renal loss, or diuretic use) owing to a decreased stretch on the baroreceptors in the great veins, aortic arch, and carotid bodies, an increased sympathetic tone to maintain systemic blood pressure generally occurs. This increased sympathetic tone, along with decreased renal perfusion secondary to intravascular volume depletion, results in increased renin and angiotensin excretion. This results in increased sodium absorption in the proximal tubules of the kidney and consequent decreased delivery of solutes to distal diluting segments, causing an impairment of renal free water excretion. There also is a concomitant increase in serum ADH (antidiuretic hormone) production that further impairs free water excretion. Because angiotensin is also a very potent stimulant of thirst, free water intake is increased, and, at the same time, water excretion is limited. Together, these changes lead to hyponatremia.
· Cerebral salt wasting (CSW) is seen with intracranial disorders (such as subarachnoid hemorrhage, carcinomatous or infectious meningitis, and metastatic carcinoma), and especially after neurologic procedures. Disruption of sympathetic neural input into the kidney, which normally promotes salt and water reabsorption in the proximal nephron segment through various indirect and direct mechanisms, might cause renal salt wasting, resulting in reduced plasma volume. Plasma renin and aldosterone levels fail to rise appropriately in patients with CSW despite a reduced plasma volume because of disruption of the sympathetic nervous system. Also, the release of natriuretic factors could play a role in the renal salt wasting seen in CSW. Volume depletion leads to an elevation of plasma vasopressin levels and impaired free water excretion. Distinguishing between CSW and syndrome of inappropriate ADH secretion (SIADH) can be challenging because there is considerable overlap in the clinical presentation. Vigorous salt replacement is required in patients with CSW, whereas fluid restriction is the treatment of choice in patients with SIADH.
· Salt-wasting nephropathy causing hypovolemic hyponatremia may rarely develop in a range of renal disorders (e.g., interstitial nephropathy, medullary cystic disease, polycystic kidney disease, partial urinary obstruction) with a low salt intake.
· Diuretics may induce hypovolemic hyponatremia. Thiazide diuretics, in contrast to loop diuretics, impair the diluting mechanism without limiting the concentrating mechanism, thereby impairing the ability to excrete a free water load. Thus, thiazides are more prone to causing hyponatremia than are loop diuretics, especially in elderly persons, who already have impaired diluting ability.
· II) Hypervolemic hypotonic hyponatremia. This is characterized by clinically detectable edema or ascites that signify an increase in total body water and sodium. Paradoxically, a decrease in the effective circulating volume, critical for tissue perfusion, stimulates the same pathophysiologic mechanism of impaired water excretion by the kidney that is observed in hypovolemic hypotonic hyponatremia. Examples include liver cirrhosis, congestive heart failure, nephrotic syndrome, and severe hypoproteinemia (albumin level < 1.5 – 2 g/dL).
· III) Normovolemic (euvolemic) hypotonic hyponatremia. This is a prevalent cause of hyponatremia in patients who are hospitalized. It is associated with nonosmotic and nonvolume-related vasopressin (ADH) secretion (i.e., SIADH) secondary to a variety of clinical conditions, including CNS disturbances, major surgery, trauma, pulmonary tumors, infection, stress, and certain medications.
· Common medications associated with SIADH are: chlorpropamide (an antidiabetic medication; potentiates renal action of ADH), carbamazepine (an antiepileptic; possesses antidiuretic property), cyclophosphamide (an alkylating agent; used for cancer chemotherapy and for autoimmune diseases; marked water retention secondary to SIADH and potentially fatal hyponatremia may ensue in selected cases), vincristine & vinblastine (cancer chemotherapy medications), amitriptyline (a tricyclic antidepressant (TCA)), haloperidol (typical antipsychotic medication), selective serotonin reuptake inhibitors (SSRIs; antidepressants; particularly have this side effect in elderly patients), and monoamine oxidase (MAO inhibitors) antidepressants. In these cases, the ability of the kidney to dilute urine in the setting of serum hypotonicity is reduced. Hyponatremia is a relatively common adverse effect of desmopressin, vasopressin (antidiuretic hormone ADH) analog that acts as a pure V2 agonist. Its common use in the treatment of central diabetes insipidus, von Willebrand disease, and nocturia in adults and of enuresis in children; it requires regular monitoring of serum sodium levels.
· The diagnostic criteria for SIADH are: normal hepatic, renal, and cardiac function - clinical euvolemia (absence of intravascular volume depletion); normal thyroid and adrenal function; hypotonic hyponatremia; urine osmolality greater than 100 mOsm/kg, generally greater than 400 – 500 mOsm/kg, with normal renal function. Urinary sodium concentrations are also typically greater than 20 mEq/L on a normal salt diet as sodium excretion will reflect dietary sodium intake. Serum uric acid levels are generally reduced due to reduced tubular uric acid reabsorption, which parallels the decrease in proximal tubular sodium reabsorption associated with central volume expansion.
· Differential diagnosis with SIADH:
· The above findings OF SIADH are also found in a renal salt wasting process. This similarity makes the differentiation between salt wasting and SIADH difficult except that in renal wasting we would expect to find a hypovolemic state. Reset osmostat is another important cause of normovolemic hypotonic hyponatremia. This may occur in elderly patients and during pregnancy. These patients regulate their serum osmolality around a reduced setpoint; however, in contrast to patients with SIADH (who also have a downward resetting of the osmotic threshold for thirst), they are able to dilute their urine in response to a water load to keep the serum osmolality around the preset low point.
· Severe hypothyroidism and adrenal insufficiency are also associated with nonosmotic vasopressin release and impaired sodium reabsorption, leading to hypotonic hyponatremia. Hyponatremia associated with cortisol deficiency, such as primary or secondary hypoadrenalism (it may present subtly and may go undiagnosed). A random cortisol level check, especially in acute illness, can be misleading if the level is normal (when it should be high). Testing for adrenal insufficiency and hypothyroidism should be part of the hyponatremic workup, as the disorders respond promptly to hormone replacement.
· Hospitalized patients who are infected with human immunodeficiency virus (HIV) have a high incidence of hyponatremia. In these patients, hyponatremia is usually due to increased release of ADH due to malignancy, to occult or symptomatic infection of the central nervous system, or to pneumonia resulting from infection with Pneumocystis jiroveci or other organisms; effective volume depletion secondary to fluid loss from the gastrointestinal tract, due primarily to infectious diarrhea; adrenal insufficiency often due to an adrenalitis, an abnormality that may be infectious in origin and may be induced by cytomegalovirus,Mycobacterium avium-intracellulare, or HIV itself.
· d) Other causes:
· i) Iatrogenic infusion of hypotonic fluids in patients after surgery. Inappropriate administration of hypotonic intravenous fluids after surgery increases the risk of developing hyponatremia in these vulnerable patients, who retain water due to the nonosmotic release of ADH, which is typically elevated for a few days after most surgical procedures. Hospital-acquired acute hyponatremia is disturbingly common also among hospitalized children and adults.
· ii) Severe malnutrition seen in weight-conscious women (low protein, high water intake diet) is a special condition in which a markedly decreased intake of solutes occurs, which limits the ability of the kidney to handle the free water. Because a mandatory solute loss of 50 – 100 mOsm/kg of urine exists, free water intake more than solute needs can produce hyponatremia.
· Another example is beer drinker's potomania because a diet consisting primarily of beer is rich in free water but solute poor.
· iii) Primary polydipsia – compulsive intake of large amounts of free water exceeding the diluting capacity of the kidneys (>20 L/day), even with a normal solute intake of 600 –900 mOsm/d, may also result in hyponatremia, but in contrast to SIADH, the urine is maximally dilute. A central defect in thirst regulation plays an important role in the pathogenesis of primary polydipsia; also different abnormalities in ADH regulation impairing free water excretion have been identified in psychotic patients. Transient stimulation of ADH release during acute psychotic episodes, an increase in the net renal response to ADH, downward resetting of the osmostat, and antipsychotic medication may contribute. Limiting water intake will rapidly raise the plasma sodium concentration as the excess water is readily excreted in dilute urine.
· iv) Ultra-endurance athletes and marathon runners. With women making up a higher percentage, the strongest single predictor is weight gain during the race correlating with an excessive fluid intake. Longer racing time and body mass index extremes are also associated with hyponatremia, whereas the composition of fluids consumed (plain water rather than sports drinks containing electrolytes) is not. Oxidization of glycogen and triglyceride during a race is associated with the production of "bound" water, which then becomes an endogenous, electrolyte-free water infusion contributing to hyponatremia induced by water ingestion more than water losses. Note: some collapsed runners are normonatremic or even hypernatremic, making blanket recommendations difficult. However, fluid intake to the point of weight gain should be avoided. Athletes should rely on thirst as their guide for fluid replacement and avoid fixed recommendations for water intake.
· v) Nonsteroidal anti-inflammatory drug (NSAID) use may increase the risk of development of hyponatremia by strenuous exercise by inhibiting prostaglandin formation. Prostaglandins have a natriuretic effect. Prostaglandin depletion increases NaCl reabsorption in the thick ascending limb of Henle and ADH action in the collecting duct, leading to impaired free water excretion.
· vi) The recreational drug of abuse ‘ecstasy’ (methylenedioxymethamphetamine, or MDMA), an amphetamine. Symptomatic and potentially fatal hyponatremia can develop with rapid onset after ingestion of the designer drug ecstasy. A marked increase in water intake via direct thirst stimulation, as well as inappropriate secretion of ADH, contributes to the hyponatremia seen with even a small amount of drug intake.
· vii) Nephrogenic syndrome of inappropriate antidiuresis (or NSIAD) is a SIADH-like clinical and laboratory picture seen in male infants who present with neurologic symptoms secondary to hyponatremia but who have undetectable plasma arginine vasopressin (AVP) levels. This hereditary disorder is secondary to mutations in the V2 vasopressin receptor, resulting in constitutive activation of the receptor with high cAMP production in the collecting duct principal cells. The current therapy of choice is fluid restriction and the use of oral urea to induce an osmotic diuresis.
· viii) Hyponatremic hypertensive syndrome, a rare condition, consists of severe hypertension associated with renal artery stenosis, hyponatremia, hypokalemia, severe thirst, and renal dysfunction characterized by natriuresis, hypercalciuria, renal glycosuria, and proteinuria. Angiotensin-mediated thirst coupled with the nonosmotic release of vasopressin provoked by angiotensin II and/or hypertensive encephalopathy are likely mechanisms for this syndrome. Sodium depletion due to pressure natriuresis and potassium depletion due to hyperaldosteronism with high plasma renin activity are also likely to play a role in the pathogenesis of hyponatremia. The abnormalities resolved with correction of the renal artery stenosis.
{kind=link}
{kind=link}
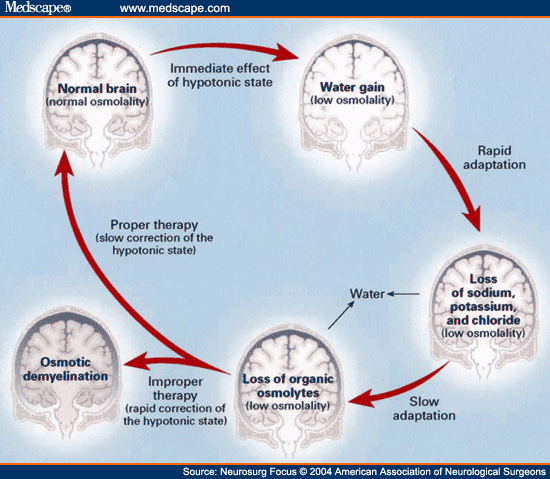
· Central pontine myelinolysis & rapid correction of hyponatremia: Central pontine myelinolysis, also known as osmotic demyelination syndrome or central pontine demyelination, is a concentrated, frequently symmetric, noninflammatory demyelination within the central basis pontis. In at least 10% of patients with central pontine myelinolysis, demyelination also occurs in extrapontine regions, including the midbrain, thalamus, basal nuclei, and cerebellum. The exact mechanism that strips the myelin sheath is unknown. Death is common. It is characterized by acute paralysis, dysphagia (difficulty swallowing), dysarthria (difficulty speaking), and other neurological symptoms.
· Maximum recovery from central pontine myelinolysis may require several months. Chronic neurologic deficits range from locked-in syndrome to spastic quadriparesis. Patients with extrapontine lesions may exhibit tremor and ataxia. Conditions predisposing patients to central pontine myelinolysis include alcoholism, liver disease, malnutrition, and hyponatremia.
· Risk factors for central pontine myelinolysis in the hyponatremic patient include serum sodium of less than 120 mEq/L for more than 48 hours; aggressive IV fluid therapy with hypertonic saline solutions; development of hypernatremia during treatment.
· Many patients who have hyponatremia that is corrected rapidly do not develop central pontine myelinolysis. Thus, other less obvious risk factors probably exist. Central pontine myelinolysis presents most commonly as a complication of treatment of patients with profound, life-threatening hyponatremia. It occurs as a consequence of a rapid rise in serum tonicity following therapy in individuals with chronic, severe hyponatremia who have made intracellular adaptations to the prevailing hypotonicity. Hyponatremia should be corrected at a rate of no more than 12 – 20 mmol/L of sodium per day to prevent central pontine myelinolysis. Central pontine myelinolysis reportedly occurs occasionally in patients who are treated for hypernatremia.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Diagnostic evaluation of hyponatremia (algorithm):
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
· Syndrome of inappropriate antidiuretic hormone secretion (SIADH): it is defined by the hyponatremia and hypo-osmolality resulting from inappropriate, continued secretion or action of the antidiuretic hormone [(ADH); also called Arginine – Vasopressin (AVP))] despite normal or increased plasma volume, which results in impaired water excretion.
· Signs & Symptoms: generally, slowly progressive hyponatremia is associated with fewer symptoms than is a rapid drop of serum sodium to the same value; signs and symptoms of acute hyponatremia do not precisely correlate with the severity or the acuity of the hyponatremia; patients may have symptoms that suggest increased secretion of ADH, such as chronic pain, symptoms from the central nervous system or pulmonary tumors or head injury, or drug use.
· Prominent physical findings may be seen only in severe or rapid-onset hyponatremia and can include: confusion, disorientation, delirium, generalized muscle weakness, myoclonus, tremor, asterixis, hyporeflexia, ataxia, dysarthria, Cheyne-Stokes respiration, pathologic reflexes, generalized seizures, and coma.
· Diagnosis: SIADH is defined by the Bartter-Schwartz criteria: hyponatremia with the corresponding hypo-osmolality; continued renal excretion of sodium; urine less than maximally dilute; absence of clinical evidence of volume depletion; absence of other causes of hyponatremia; and correction of hyponatremia by fluid restriction.
· Causes of SIADH:
· a) Nervous system disorders: acute psychosis; acute intermittent porphyria; brain abscess; cavernous sinus thrombosis; cerebellar and cerebral atrophy; cerebrovascular accident; CNS (central nervous system) lupus; delirium tremens; encephalitis (viral or bacterial); epilepsy; Guillain-Barré syndrome; head trauma; herpes zoster (on chest wall); hydrocephalus; hypoxic-ischemic encephalopathy; meningitis (viral, bacterial, tuberculous, and fungal); midfacial hypoplasia; multiple sclerosis (MS); perinatal hypoxia; Rocky Mountain spotted fever; schizophrenia; Shy-Drager syndrome (*); subarachnoid hemorrhage; subdural hematoma; ventriculoatrial shunt obstruction; Wernicke encephalopathy (from thiamine (vitamin B1) deficiency e.g. on malnourished alcoholics).
· b) Neoplasia disorders: pulmonary (lung carcinoma and mesothelioma); gastrointestinal (carcinomas of the duodenum, pancreas, and colon); genitourinary (adrenocortical carcinoma; carcinomas of cervix, ureter/bladder, and prostate; and ovarian tumors); other [brain tumors, carcinoid tumors, Ewing sarcoma, leukemia, lymphoma, nasopharyngeal carcinoma, neuroblastoma (olfactory), and thymoma].
· c) Pulmonary disorders: acute bronchitis/bronchiolitis; acute respiratory failure; aspergillosis (cavitary lesions); asthma; atelectasis; bacterial pneumonia; chronic obstructive lung disease (COPD); cystic fibrosis; emphysema; empyema; pneumonia [viral, bacterial (mycoplasmal), fungal]; pneumothorax; positive pressure ventilation; pulmonary abscess; pulmonary fibrosis; sarcoidosis; tuberculosis (TB); viral pneumonia.
· d) Drugs that stimulate AVP release: acetylcholine; antineoplastic agents (adenine arabinoside, cyclophosphamide, ifosfamide, vincristine, vinblastine); barbiturates; bromocriptine; carbachol; chlorpropamide; clofibrate; cyclopropane; dibenzazepines (eg, carbamazepine, oxcarbazepine); halothane; haloperidol; histamine; isoproterenol; lorcainide; opiates e.g. morphine; nicotine (inhaled tobacco); nitrous oxide (NO); phenothiazines (eg, thioridazine); thiopental; MAO inhibitors (eg, tranylcypromine); tricyclic antidepressants (TCAs eg, amitriptyline, desipramine).
· e) Drugs that potentiate the effects of AVP action (primarily facilitates peripheral action of ADH): clofibrate; griseofulvin; hypoglycemic agents (metformin, phenformin, tolbutamide); oxytocin (large doses); prostaglandin synthetase inhibitors (inhibit renal PGE2 synthesis; indomethacin, aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs); theophylline; triiodothyronine; vasopressin analogs (eg, AVP, DDAVP).
· f) Drugs with an uncertain mechanism: antineoplastic agents (cisplatin, melphalan, methotrexate, imatinib); ciprofloxacin; clomipramine; ecstasy; phenoxybenzamine; sodium valproate; SSRIs (e.g., sertraline, fluoxetine, paroxetine); and thiothixene.
· Note: The list of drugs that can induce SIADH is long.
· SIADH has been reported as an adverse effect of multiple psychotropic medications.
· Many chemotherapeutic drugs cause nausea, which is a powerful stimulus of vasopressin secretion.
· SIADH is also a leading cause of hyponatremia in children following chemotherapy or stem cell transplantation.
· g) Miscellaneous causes: exercise-induced hyponatremia; giant cell arteritis; HIV infection (hyponatremia has been reported in as many as 40% of adult patients with HIV infection; patients with AIDS can have many potential causes for increased ADH secretion, including volume depletion and infection of the lungs and the CNS); idiopathic.
· (*) Multiple-system atrophy (MSA) is a degenerative neurological disorder associated with the degeneration of nerve cells in specific areas of the brain. This cell degeneration causes problems with movement, balance, and autonomic functions of the body such as bladder control or blood-pressure regulation. The cause of MSA is unknown, and no specific risk factors have been identified. About 55% of cases occur in men, with the typical age of onset in the late 50s to early 60s. It often presents some of the same symptoms as Parkinson’s disease. However, MSA patients generally show minimal if any response to the dopamine medications used for Parkinson's. When autonomic failure predominates, the term Shy–Drager syndrome is sometimes used, although this term is no longer current, given the terminology changes.
· Νote: Vasopressin, also known as arginine vasopressin (AVP), antidiuretic hormone (ADH), or argipressin, is a neurohypophysial hormone found in most mammals. Its two primary functions are to retain water in the body and to constrict blood vessels. Vasopressin regulates the body's retention of water by acting to increase water reabsorption in the kidney's collecting ducts, the tubules which receive the very dilute urine produced by the functional unit of the kidney, the nephrons. Vasopressin is a peptide hormone that increases water permeability of the kidney's collecting duct and distal convoluted tubule. It also increases peripheral vascular resistance, which in turn increases arterial blood pressure (BP). It plays a key role in homeostasis (regulation), by the regulation of water, glucose, and salts in the blood.
{kind=link}
· SIADH diagnostic algorithm:
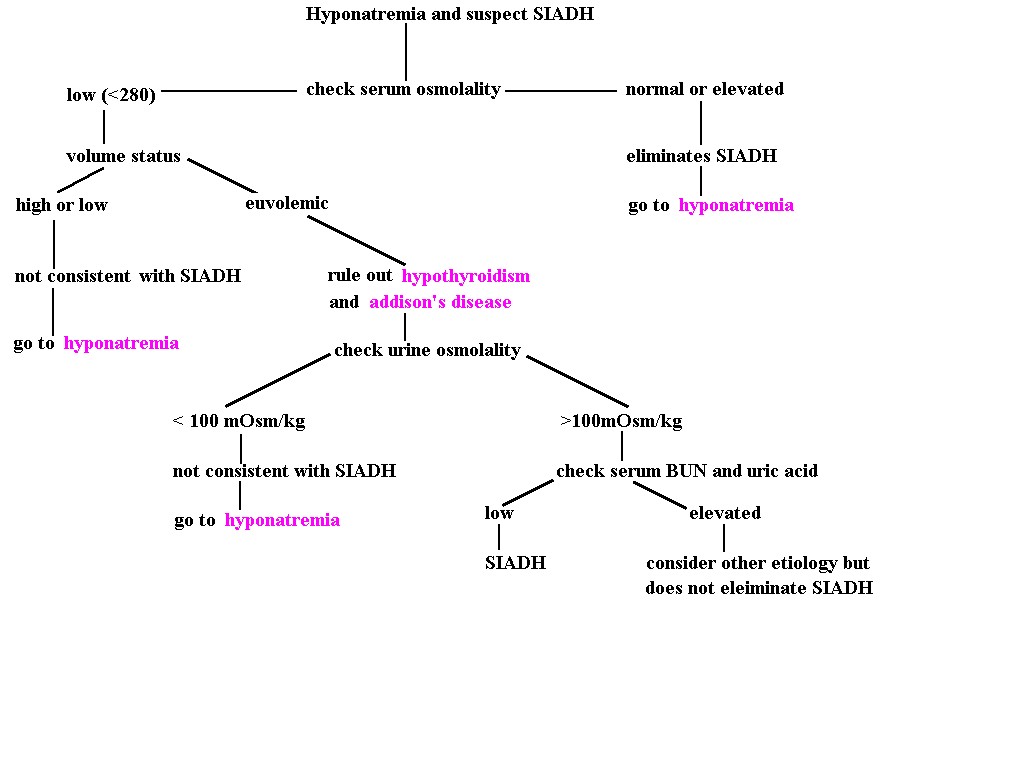{kind=link}
· Fractional excretion of uric acid (FEUa) & SIADH: Fractional excretion of uric acid (FEUa) is measured with the formula FEuric acid (FEUa)%= (Urine uric acid x plasma Creatinine) / (Plasma uric acid x Urine Creatinine)
· It is used to differentiate between renal and prerenal acute kidney injury (AKI).
· FEUa > 10% (>0.1 fraction) indicates SIADH or renal causes (including acute tubular necrosis (ATN)).
· FEUa < 10% (<0.1 fraction) indicates prerenal causes.
· Fractional excretion of uric acid is not affected by a loop or thiazide diuretics.
· In a study, 86 consecutive hyponatremic patients (serum Na <130 mmol/liter) that were classified based on their history, clinical evaluation, osmolality, and saline response to isotonic saline into a SIAD (syndrome of inappropriate antidiuresis) and a non-SIAD group. A total of 31 patients (36%) had a diagnosis of SIAD, and 55 (64%) were classified as non-SIAD. There were 57 patients (68%) who were on diuretics. In the absence of diuretic therapy, SIAD was accurately diagnosed using U-Na (urinary sodium (Na) excretion). However, in patients on diuretics, the diagnosis was unreliable. There, FE-UA (fractional excretion of uric acid) performed best compared with all other markers tested, resulting in a positive predictive value of 100% if a cutoff value of 12% was used. The study concluded that FE-UA allows the diagnosis of SIAD with excellent specificity. Combining the information on U-Na (urinary sodium (Na) excretion) and FE-UA (fractional excretion of uric acid) leads to a very high diagnostic accuracy in hyponatremic patients with and without diuretic treatment.
· Hypernatremia: is a common electrolyte problem and is defined as a rise in serum sodium concentration to a value exceeding 145 mmol/L. It is strictly defined as a hyperosmolar condition caused by a decrease in total body water (TBW) relative to electrolyte content. Hypernatremia actually is a ‘water problem,’ not a problem of sodium homeostasis. Patients developing hypernatremia outside of the hospital are generally elderly people who are mentally and physically impaired, often with an acute infection.
· Signs & symptoms: symptoms can be related to volume deficit and/or hypertonicity and shrinkage of brain cells, which can tear cerebral blood vessels in severe cases, leading to cerebral hemorrhage. Several risk factors exist for hypernatremia.
· The most significant risk factor is age older than 65 years. Moreover, mental or physical disability may result in impaired thirst sensation, a reduced ability to express thirst, and/or decreased access to water. The most prominent concurrent factor is inadequate fluid intake. But, developing hypernatremia is virtually impossible if the thirst response is intact and water is available. Typically, an increase in osmolality of just 1 –2% stimulates thirst, as do hypovolemia and hypotension.
· Characteristics:
· a) Cognitive dysfunction and symptoms associated with neuronal cell shrinkage – related symptoms: lethargy, obtundation, confusion, abnormal speech, irritability, seizures, nystagmus, myoclonic jerks.
· b) Dehydration or clinical signs of volume depletion – related symptoms: οrthostatic blood pressure changes, tachycardia, oliguria, dry oral mucosa, abnormal skin turgor, dry axillae.
· c) Other clinical findings – related symptoms: weight loss, generalized weakness.
· Causes of hypernatremia: hypernatremia can, in a simplified view, be classified by the concurrent water loss or electrolyte gain and on corresponding changes in extracellular fluid volume:
· I) Loss of hypotonic fluid (loss of water more than electrolytes). Patients who lose hypotonic fluid have a deficit in free water and electrolytes (low total body sodium and potassium) and have decreased extracellular volume. In these patients, hypovolemia may be more life-threatening than hypertonicity. When physical evidence of hypovolemia is present, fluid resuscitation with normal saline is the first step in therapy.
· Renal hypotonic fluid loss results from causes that interfere with the ability of the kidney to concentrate the urine or osmotic diuresis, such as:
· a) Diuretic drugs: loop and thiazide diuretics.
· b) Osmotic diuresis: hyperglycemia, mannitol, urea (high-protein tube feeding).
· c) Post-obstructive diuresis.
· d) Diuretic phase of acute tubular necrosis.
· Non-renal hypotonic fluid loss can result from:
· a) GI (gastrointestinal) causes: vomiting, diarrhea, lactulose, cathartics, nasogastric suction, gastrointestinal fluid drains, and fistulas.
· b) Cutaneous (related to the skin): sweating (extreme sports, marathon runners), burn injuries.
· II) Pure-water deficits. Patients with pure-water deficits in the majority of cases have a normal extracellular volume with normal total body sodium and potassium. This condition most commonly develops when impaired intake is combined with increased insensible (e.g., respiratory) or renal water losses. The free-water loss may also result from an inability of the kidney to concentrate the urine. The cause of that can be either from the failure of the hypothalamic-pituitary axis to synthesize or release adequate amounts of AVP (central diabetes insipidus) or a lack of responsiveness of the kidney to AVP (nephrogenic diabetes insipidus). Patients with diabetes insipidus and intact thirst mechanisms most often present with normal plasma osmolality and serum sodium, but with symptoms of polyuria and polydipsia.
· Water intake less than insensible losses may result from:
· a) Lack of access to water: incarceration (imprisonment), restraints, intubation, immobilization.
· b) Altered mental status: medications, diseases.
· c) Neurologic disease: dementia, impaired motor function.
· d) Abnormal thirst: e.g., geriatric hypodipsia; osmoreceptor dysfunction (reset of the osmotic threshold); injury to the thirst centers by any lesions to the hypothalamus (including metastasis, granulomatous diseases, vascular abnormalities, and trauma); autoantibodies to the sodium-level sensor in the brain.
· e) Loss of water through the respiratory tract.
· III) Vasopressin (AVP) deficiency (diabetes insipidus).
· Central diabetes insipidus can be caused by any pathologic process that destroys the anatomic structures of the hypothalamic-pituitary axis involved in AVP production and secretion.
· Causes include:
· a) Pituitary injury: post-traumatic, neurosurgical, hemorrhage, ischemia (Sheehan’s syndrome (*)), idiopathic/ autoimmune, lymphocytic hypophysitis, IgG4-related disease.
· (*) Sheehan’s syndrome: hypopituitarism (decreased functioning of the pituitary gland) caused by ischemic necrosis due to blood loss and hypovolemic shock during and after childbirth.
· b) Tumors: craniopharyngioma, pinealoma, meningioma, germinoma, lymphoma, metastatic disease, cysts.
· c) Aneurysms: mainly anterior communicating.
· d) Inflammatory states and granulomatous disease: acute meningitis/encephalitis, Langerhans cell histiocytosis, neurosarcoidosis, tuberculosis.
· e) Drugs: ethanol (transient), phenytoin (antiepileptic).
· f) Genetic: neurophysin II (AVP carrier protein) gene defect.
· IV) Nephrogenic diabetes insipidus (decreased responsiveness of the kidney to vasopressin).
· Causes include:
· a) Genetic: V2-receptor defects, aquaporin defects (AQP2 and AQP1); 90% byAVPR2 mutations (X-liked recessive), AQP2 gene mutation.
· b) Structural: urinary tract obstruction, papillary necrosis, sickle-cell nephropathy.
· c)Tubulointerstitial disease: medullary cystic disease, polycystic kidney disease, nephrocalcinosis, Sjogren’s syndrome, lupus, analgesic-abuse nephropathy, sarcoidosis, M-protein disease, cystinosis, nephronophthisis.
· d) Other causes: distal renal tubular acidosis, Bartter syndrome, apparent mineralocorticoid excess.
· e) Electrolyte disorders: hypercalcemia, hypokalemia.
· f) Any prolonged state of severe polyuria.
· g) Medications:
· i) Medications that induce nephrogenic diabetes insipidus include: lithium (40% of patients; lithium is used for bipolar disorder); amphotericin B (for fungal infection); demeclocycline (a tetracycline antibiotic used for bacterial infections and for SIADH); dopamine; ofloxacin (an antibiotic); orlistat (a lipase – inhibitor; used on treating obesity); and ifosfamide (a nitrogen mustard alkylating agent used for cancer chemotherapy).
· ii) Medications that possibly cause nephrogenic diabetes insipidus to include contrast agents, cyclophosphamide, cidofovir, ethanol, foscarnet, indinavir, libenzapril, mesalazine, methoxyflurane, pimozide, rifampin, streptozocin, tenofovir, triamterene hydrochloride, and colchicine.
· V) Adipsic diabetes insipidus (central diabetes insipidus with deficient thirst). It is caused by a combination of damage to the osmoreceptors regulating thirst sensation and central diabetes insipidus.
· Causes include:
· a) Congenital conditions: septo-optic dysplasia, germinoma.
· b) Vascular causes: anterior communicating artery aneurysm clipping/rupture.
· c) Other causes: craniopharyngioma, pinealoma, Langerhans cell histiocytosis, neurosarcoidosis, head trauma, cytomegalovirus (CMV) encephalitis.
· VI) Gestational diabetes insipidus. Here AVP is rapidly degraded by a high circulating level of oxytocinase/vasopressinase. It is a rare condition because increased AVP secretion will compensate for the increased rate of degradation. Gestational diabetes insipidus occurs only in combination with impaired AVP production.
· VII) Hypertonic sodium gain. Patients with hypertonic sodium gain have a high total-body sodium and an extracellular volume overload (rare, mostly iatrogenic). When thirst and renal function are intact, this condition is transient.
· Causes include:
· a) Administration of hypertonic electrolyte solutions, e.g., sodium bicarbonate solutions, hypertonic alimentation solutions, normal saline with or without potassium supplements.
· b) Sodium ingestion: NaCl tablets, seawater ingestion.
· c) Sodium modeling in hemodialysis.
· VIII) Water shift (transient). Water shifts into muscle cells during extreme exercise or seizures because of increased intracellular osmoles. In clinical practice, a combination of the two may be present, e.g., an intubated patient in the ICU develops hypernatremia due to hypertonic sodium gain caused by normal saline volume resuscitation and, also, increased free water excretion due to recovering renal failure and/or osmotic urea-diuresis caused by high-protein tube feeding.
· Diabetes insipidus (DI): is a condition characterized by excessive thirst and excretion of large amounts of severely diluted urine, with reduction of fluid intake having no effect on the concentration of the urine. There are different types of DI, each with a different set of causes. The most common type in humans is the neurological form, called central diabetes insipidus (CDI), which involves a deficiency of arginine vasopressin (AVP), also known as antidiuretic hormone (ADH). The second common type of DI is nephrogenic diabetes insipidus (NDI), which is due to kidney or nephron dysfunction caused by an insensitivity of the kidneys or nephrons (the basic structural and functional unit of the kidney) to ADH.
· DI can also be gestational or caused by alcohol or some types of drug abuse.
· Although they have a common name, diabetes mellitus and diabetes insipidus are two entirely separate conditions with independent mechanisms. Both cause massive amounts of urine to be produced (polyuria). However, diabetes insipidus is either a problem with the production of antidiuretic hormone (central diabetes insipidus) or kidney's response to antidiuretic hormone (nephrogenic diabetes insipidus), whereas diabetes mellitus causes polyuria via osmotic diuresis, due to the high blood sugar leaking into the urine and taking excess water along with it.
· The incidence of diabetes insipidus in the general population is 3 in 100,000.
· Diagnosis: to distinguish DI from other causes of excess urination, blood glucose levels, bicarbonate levels, and calcium levels need to be tested. Measurement of blood electrolytes can reveal a high sodium level (hypernatremia) as dehydration develops. Urinalysis shows dilute urine with low specific gravity. Urine osmolarity and electrolyte levels are typically low.
· A fluid deprivation test is a way of distinguishing DI from other causes of excessive urination. It is also used to help determine what DI is caused by a defect in ADH production or an error in the kidneys' response to ADH. This test measures the changes in body weight, urine output, and urine composition when fluids are withheld to induce dehydration. The body's normal response to dehydration is to conserve water by concentrating the urine.
· Those with DI continue to urinate large amounts of dilute urine in spite of water deprivation.
· In primary polydipsia, the urine osmolality should increase and stabilize at above 280 Osm/kg with fluid restriction, while stabilization at a lower level indicates diabetes insipidus. Stabilization in this test means that an increase in urine osmolality is less than 30 Osm/kg per hour for at least 3 hours. Sometimes measuring blood levels of ADH toward the end of this test is also necessary, but is more time-consuming.
· To distinguish between the main forms, desmopressin (DDAVP, a synthetic analog of ADH) stimulation is also used. Desmopressin can be taken by injection, a nasal spray, or a tablet. While taking desmopressin, a patient should drink fluids or water only when thirsty and not at other times, as this can lead to sudden fluid accumulation in the central nervous system.
· If desmopressin reduces urine output and increases urine osmolarity, the hypothalamic production of ADH is deficient, and the kidney normally responds to exogenous vasopressin (desmopressin).
· If the DI is due to kidney pathology, desmopressin does not change either urine output or osmolarity, since the endogenous vasopressin levels are already high.
· If central DI is suspected, testing of other hormones of the pituitary and also MRI, particularly a pituitary MRI, is necessary to discover if a disease process (such as a prolactinoma, histiocytosis, syphilis, tuberculosis, other tumors, granulomata) is affecting pituitary function. Most people with this form have either experienced past head trauma or have stopped ADH production for an unknown reason.
· Νote: Vasopressin, also known as arginine vasopressin (AVP), antidiuretic hormone (ADH), or argipressin, is a neurohypophysial hormone found in most mammals. Its two primary functions are to retain water in the body and to constrict blood vessels. Vasopressin regulates the body's retention of water by acting to increase water reabsorption in the kidney's collecting ducts, the tubules which receive the very dilute urine produced by the functional unit of the kidney, the nephrons. Vasopressin is a peptide hormone that increases water permeability of the kidney's collecting duct and distal convoluted tubule. It also increases peripheral vascular resistance, which in turn increases arterial blood pressure (BP). It plays a crucial role in homeostasis (regulation), by the control of water, glucose, and salts in the blood.
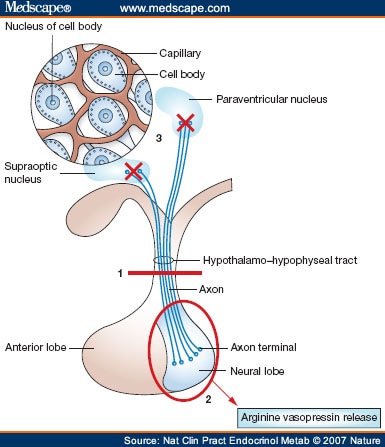{kind=link}
· Polyuria & polydipsia algorithm:
{kind=link}
· Diagnostic evaluation of hypernatremia (algorithm):
{kind=link}
{kind=link}
{kind=link}
· ECFV: extracellular fluid volume (see https://en.wikipedia.org/wiki/Extracellular_fluid )
· Sodium urine: The reference range is 43 – 217 mEq/24 hours. Specimen requirement is a 24-hour urine collection in a container without preservatives. The specimen should be refrigerated during and after the collection.
· Ιnterpretation:
· 1) On patients with hyponatremia:
· I) Hypovolemia:
· a) Urinary sodium > 20 mEq/L: Causes: renal losses due to diuretics, aldosterone deficiency, salt-losing nephropathy, osmotic diuresis, ketonuria, RTA (renal tubular acidosis).
· b) Urinary sodium < 20 mEq/L: Causes: increased renal losses due to vomiting, diarrhea, third spacing of fluids from burns, pancreatitis, trauma.
· II) Euvolemia: Urinary sodium > 20 mEq/L: Causes: cortisol deficiency, hypothyroidism, stress, drugs, SIADH.
· II) Hypervolemia:
· a) Urinary sodium > 20 mEq/L: Causes: acute or chronic renal failure.
· b) Urinary sodium < 20 mEq/L: Causes: Nephrotic syndrome, cirrhosis, cardiac failure.
· 2) On patients with hypernatremia:
· I) Hypovolemia:
· a) Urinary sodium < 20 mEq/L: Causes: excess sweating, burns, diarrhea, fistulas.
· b) Urinary sodium > 20 mEq/L: Causes: renal disease, urinary tract obstruction, osmotic or loop diuretics.
· II) Euvolemia: Urinary sodium: variable. Causes: diabetes insipidus, hypodipsia, insensible losses, respiratory/ dermal (skin) losses.
· III) Hypervolemia: a) Urinary sodium > 20 mEq/L: Causes: primary hyperaldosteronism, Cushing’s syndrome, hypertonic dialysis, hypertonic sodium bicarbonate, sodium chloride tablets.
· 3) Urinary sodium and causes of acute renal failure (ARF):
· a) Urinary sodium < 20 mEq/L: prerenal causes of acute renal failure.
· b) Urinary sodium > 40 mEq/L: renal causes of acute renal failure.
· Note: FENa% (fractional excretion of sodium (see below)) and urine sodium concentration are difficult to interpret with concurrent diuretic therapy. The fractional excretion of urea (FEurea) (see below) may be most useful in this setting, although inconsistent results have been reported. In general, the FEurea is between 50 – 65% in ATN (acute tubular necrosis) and usually < 35% in prerenal disease.
· Fluid balance:
· Hypovolaemia (oligemia or shock) is a state of decreased blood volume; more specifically, reduce in the volume of blood plasma.
· Euvolemia is the state of normal body fluid volume.
· Hypervolemia (fluid overload) is the medical condition where there is too much fluid in the blood. Fluid volume excess in the intravascular compartment occurs due to an increase in total body sodium content and a consequent increase in extracellular body water. The mechanism usually stems from compromised regulatory mechanisms for sodium handling as seen in congestive heart failure (CHF), kidney failure, and liver failure. It may also be caused by excessive intake of sodium from foods, intravenous (IV) solutions and blood transfusions, medications, or diagnostic contrast dyes.
· Fractional excretion of sodium (FENa): it measures the % of filtered sodium that is excreted in the urine. The FENa is the most accurate screening test to help differentiate between prerenal disease and acute tubular necrosis (ATN), the two most common causes of acute kidney injury. FENa is generally more accurate than the urine sodium concentration among patients with acute kidney injury. It is measured with the formula FENa%= (Urine Na x Plasma Creatinine) / (Plasma Na x Urine Creatinine)
· Note: FENA is useful in AKI (acute kidney injury) only with the presence of oliguria.
· Interpretation of fractional excretion of sodium (FENa) (on advanced renal failure):
· a) A value of FENa < 1% suggests prerenal disease (e.g., congestive heart failure (CHF) or dehydration), where the reabsorption of almost all of the filtered sodium represents an appropriate response to decreased renal perfusion. In addition to prerenal disease, FENa can be < 1% in patients with post-ischemic ATN, burns, ATN superimposed upon chronic prerenal disease, kidney injury due to radiocontrast media or heme pigments, rhabdomyolysis, acute glomerulonephritis (GN) or vasculitis, some cases of acute interstitial nephritis and rarely on acute urinary tract obstruction. Other causes include acute partial urinary tract obstruction and also hepatorenal syndrome.
· b) A value of FENa > 2% usually indicates acute tubular necrosis (ATN) (intrinsic renal cause).
· c) A value between 1 – 2% may be seen with either disorder, i.e., either prerenal or ATN. FENa > 1% also occurs on severe bilateral obstruction of the urinary drainage.
· The relatively high FENa in ATN can be due to one or both of the following factors: inappropriate sodium wasting due to tubular damage and an appropriate response to volume expansion. The former is most likely to be important early in the disease when nephrons that are still filtering have impaired tubular function. In comparison, volume expansion may be more critical in patients with established ATN and normal tissue perfusion. These cut-off values apply only to patients with advanced renal failure. Interpretation of results from patients in non-oliguric states, those with glomerulonephritis, and those receiving or ingesting diuretics can lead to an erroneous diagnosis.
· FENa can be a valuable test for helping to detect extreme renal avidity for sodium in conditions such as hepatorenal syndrome. In the presence of liver disease, FENa can be less than 1% in the presence of ATN. On the other hand, because administration of diuretics may cause the FENa to be higher than 1%, these findings cannot be used as the sole indicators in AKI.
· FENa and urine sodium concentration are difficult to interpret with concurrent diuretic therapy. The fractional excretion of urea (FEurea) (see below) may be most useful in this setting, although inconsistent results have been reported. In general, the FEurea is between 50 – 65% in acute tubular necrosis (ATN) and usually < 35% in prerenal disease.
· Fractional excretion of sodium (FENa) calculator:
· Fractional excretion of urea (FEurea): fractional excretion of sodium (FENa) and urine sodium concentration are difficult to interpret in patients treated with diuretics, because the ensuing natriuresis raises FENa, even in patients with a prerenal disease. In patients treated with diuretics, calculating the fractional excretion of urea (FEurea) may be helpful, because urea is primarily absorbed in the proximal tubule. It is measured with the formula
FEurea%= (Urine urea x plasma Creatinine) / (Plasma urea x Urine Creatinine)
FEurea%= (Urine urea x plasma Creatinine) / (Plasma urea x Urine Creatinine)
· The accuracy of FEurea depends on intact proximal function because fractional excretion of urea increases when proximal reabsorption is reduced.
· Interpretation of fractional excretion of urea (FEurea):
· a) In ATN (acute tubular necrosis) typically FEurea is 50 – 65% ATN.
· b) In prerenal disease FEurea is < 35%.
· Although still controversial, the preponderance of the evidence suggests that FEurea is more accurate than FENa in patients receiving diuretics.
· Fractional excretion of urea (FEurea) calculator:
· Phosphorus/ Phosphate (PO4–3): is a charged particle (ion) that contains the mineral phosphorus. The body needs phosphorus to build and repair bones and teeth, help nerves function, and make muscles contract. About 85% of the phosphorus contained in phosphate is found in bones. The rest of it is stored in tissues throughout the body. Phosphate is critical for a remarkably wide array of cellular processes. It is one of the major components of the skeleton, providing mineral strength to the bone. Phosphate is an integral component of the nucleic acids that comprise DNA and RNA. Phosphate bonds of ATP carry the energy required for all cellular functions. It also functions as a buffer in bone, serum, and urine. The addition and deletion of phosphate groups to enzymes and proteins are common mechanisms for the regulation of their activity. The kidneys help control the amount of phosphate in the blood. Extra phosphate is filtered by the kidneys and passes out of the body in the urine.
· A high level of phosphate in the blood is usually caused by a kidney problem.
· The amount of phosphate in the blood affects the level of calcium in the blood. Calcium and phosphate in the body react in opposite ways: as blood calcium levels rise, phosphate levels fall.
· Parathyroid hormone (PTH) regulates the levels of calcium and phosphorus in the blood.
· When the phosphorus level is measured, a vitamin D, and sometimes a PTH level, is measured at the same time. Vitamin D is needed for the body to take in phosphate. The relation between calcium and phosphate may be disrupted by some diseases or infections. For this reason, phosphate and calcium levels are usually measured at the same time.
· A test to measure phosphate in the blood may be done to:
· a) Check phosphate levels on people who have kidney disease or bone disease.
· b) To help find problems with certain glands, such as the parathyroid glands.
· c) To find a reason for abnormal vitamin D levels.
· While phosphorus tests are most commonly performed on blood samples, phosphorus is sometimes measured in urine samples to monitor its elimination by the kidneys.
· Since mildly abnormal phosphorus levels usually cause no symptoms, phosphorus testing is typically performed in follow-up to an abnormal calcium test, and/or when symptoms of abnormal calcium such as fatigue, muscle weakness, cramping, or bone problems are present. Phosphorus testing may also be ordered along with other tests when symptoms suggest kidney and gastrointestinal disorders. When conditions causing abnormal phosphorus and/or calcium levels are found, testing for both may be ordered at regular intervals to monitor treatment effectiveness. Also, on people with diabetes mellitus or signs of an acid-base imbalance, a doctor may sometimes monitor phosphorus levels.
{kind=link}
{kind=link}
{kind=link}
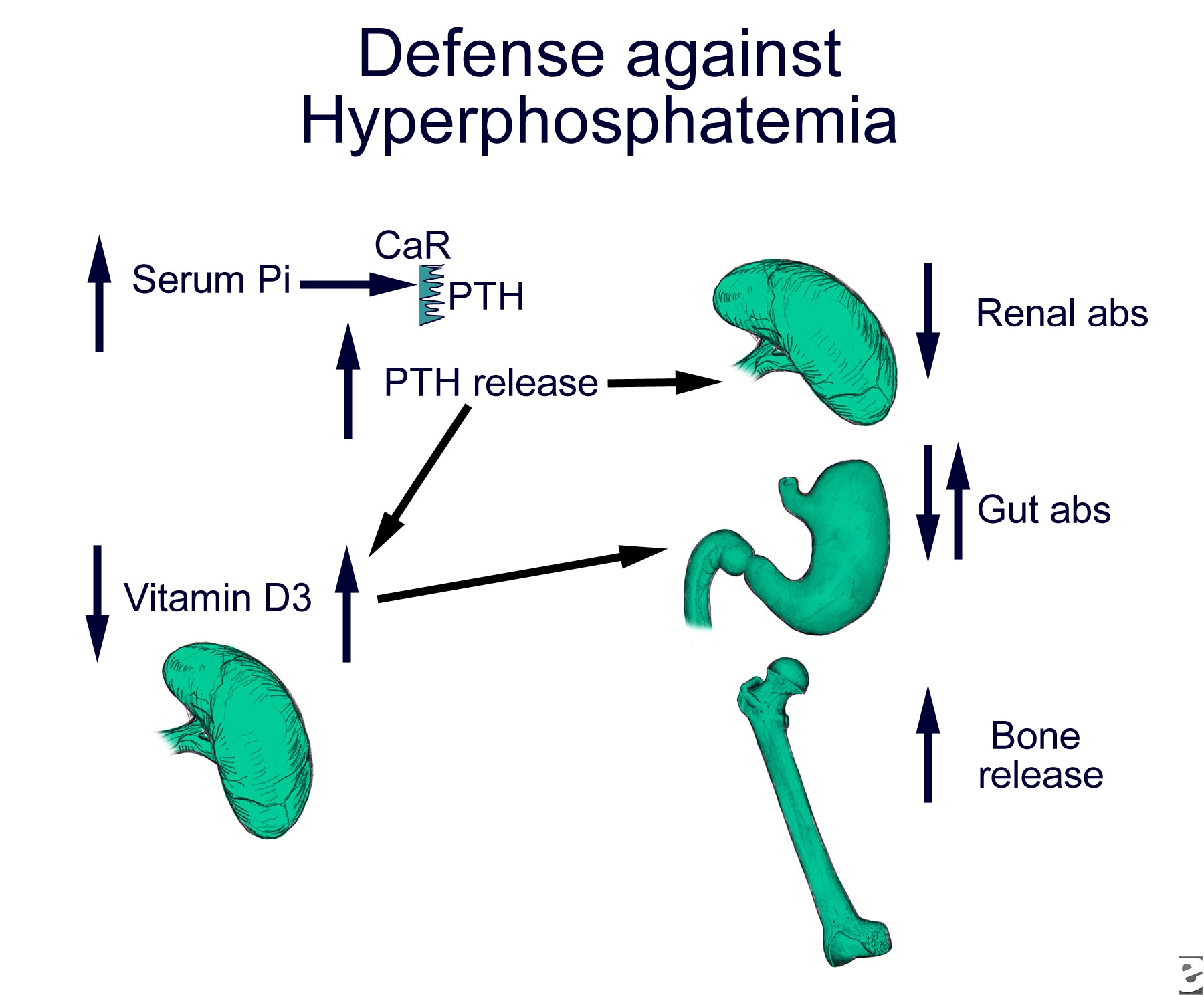{kind=link}
{kind=link}
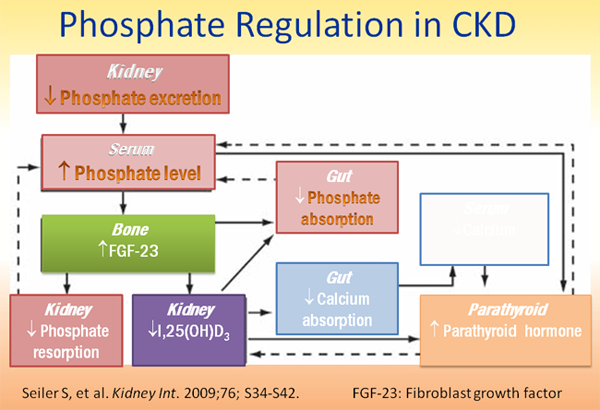{kind=link}
· Hypophosphatemia: Hypophosphatemia is low levels of phosphorus in the blood. It is defined as a phosphate level of less than 2.5 mg/dL (0.8 mmol/L).
· Signs & symptoms: no physical signs are specific for hypophosphatemia. In fact, physical signs of mild hypophosphatemia are generally absent. Chronic hypophosphatemia can be associated with short stature and evidence of rickets (in children), with bowing of the legs, when caused by one of the genetically transmitted phosphate wasting disorders. In adults, chronic hypophosphatemia is more commonly associated with bone pain upon palpation.
· Severe acute hypophosphatemia can have a variety of signs, including disorientation, seizures, focal neurologic findings, evidence of heart failure, and muscle pain. Myocardial contractility may be impaired from the depletion of adenosine triphosphate (ATP), and respiratory failure due to the weakness of the diaphragm has been described. The reduction in cardiac output may become clinically significant, leading to congestive heart failure, when the plasma phosphate concentration falls to 1.0 mg/dL (0.32 mmol/L).
· Acute hypophosphatemia superimposed upon preexisting severe phosphate depletion can lead to rhabdomyolysis. Although creatine phosphokinase (CPK) elevations are fairly common in hypophosphatemia, clinically significant rhabdomyolysis has been described almost exclusively in alcoholics and in patients receiving hyperalimentation without phosphate supplementation.
· Causes of hypophosphatemia:
· I) Inadequate intake in the diet or from poor intestinal absorption. Hypophosphatemia due to inadequate intake is uncommon but should be strongly considered in certain patient populations such as:
· a) People who have had a prolonged poor intake of phosphate: develop true phosphate deficiency.
· b) Persons with alcoholism who ingest an inadequate diet; serum phosphate levels may be within reference ranges upon admission to the hospital, but refeeding stimulates cellular uptake and results in profound hypophosphatemia.
· c) Critically ill patients receiving a parenteral diet deficient in phosphate: they may suddenly become hypophosphatemic as their catabolic condition resolves and they become more anabolic.
· d) People with eating disorders or dietary deficiencies due to socioeconomic, dental, or swallowing difficulties: may also become hypophosphatemic when fed an adequate diet.
· e) People with malabsorption of intestinal phosphate.
· f) People who ingest large quantities of antacids: can become hypophosphatemic because of phosphate binding by the antacids, resulting in poor intestinal absorption.
· g) People with primary intestinal disorders, such as Crohn's disease or celiac sprue that can limit phosphate absorption.
· h) Steatorrhea or chronic diarrhea can cause mild-to-moderate hypophosphatemia due to decreased phosphate absorption from the gut and renal phosphate wasting; the latter is caused by secondary hyperparathyroidism induced by concomitant vitamin D deficiency.
· i) Vitamin D deficiency causes hypophosphatemia by limiting intestinal and renal phosphate absorption.
· II) Excessive losses. Phosphate wasting can result from genetic or acquired renal disorders.
· 1) Genetic disorders. The genetic disorders generally manifest in infancy, when the children exhibit short stature and bone deformities. Genetic disorders that cause phosphate wasting include:
· a) X-linked hypophosphatemic rickets. It is characterized by short stature, radiographic evidence of rickets, and bone pain. Patients may also have calcification of tendons, cranial abnormalities, and spinal stenosis. In addition to hypophosphatemia, these patients have relatively low levels of 1,25 dihydroxy vitamin D-3, levels that are inappropriately low for the degree of hypophosphatemia. The defective gene is PHEX, which encodes for a membrane-bound neutral endopeptidase.
· b) Autosomal dominant hypophosphatemic rickets. It has similar manifestations, with hypophosphatemia, clinical rickets, and inappropriately low levels of 1,25 dihydroxy vitamin D-3. The cause of this disorder is thought to be mutations of the FGF23 gene that result in resistance to degradation, persistently high circulating levels of FGF23, and subsequent phosphaturia.
· c) Hereditary hypophosphatemic rickets with hypercalciuria. It is a rare disorder characterized by hypophosphatemia, phosphate wasting, hypercalciuria, bone pain, muscle weakness, and high levels of 1,25 dihydroxy vitamin D-3. The cause of this disorder is an inactivating mutation in the type 2c sodium-phosphate cotransporter.
· d) Vitamin D–resistant rickets. It is an autosomal recessive disorder. In type I, the defect is in renal 1-alpha-hydroxylation. Type II is characterized by end-organ resistance to the effects of 1,25 dihydroxy vitamin D-3. These patients present in childhood with hypocalcemia, hypophosphatemia, hyperparathyroidism, rickets, bone pain, muscle weakness, and alopecia (hair loss). The disease is caused by mutations in the vitamin D receptor that prevent normal responsiveness to circulating vitamin D.
· e) Mutations in the type 2a sodium-phosphate cotransporter. Mutations in the type 2a sodium-phosphate cotransporter have been reported in some patients with hypophosphatemia and inappropriate urinary phosphate wasting associated with nephrolithiasis and/or osteoporosis.
· f) Fibrous dysplasia/McCune-Albright syndrome. Rarely, significant renal phosphate wasting is observed in patients with fibrous dysplasia/McCune-Albright syndrome, disorders that result from mutations in the alpha subunit of the stimulatory G protein. Excess production of FGF23 has been found in some of these patients.
· 2) Acquired phosphate-wasting syndromes:
· a) Simple vitamin D deficiency results in hypophosphatemia, at least in part, from renal wasting. Vitamin D deficiency can result from several mechanisms, including poor oral intake, lack of sun exposure, drug-induced hypermetabolism of vitamin D precursors in the liver, or loss of vitamin D binding protein in the urine in persons with nephrotic syndrome. The loss of normal bone mineralization produces rickets in children and osteomalacia in adults.
· b) Primary hyperparathyroidism: causes renal phosphate wasting.
· c) Heavy metal intoxication and paraproteinemias can cause global proximal renal tubule dysfunction. These patients have hypophosphatemia along with type II renal tubular acidosis, renal glycosuria, aminoaciduria, and hypouricemia (Fanconi syndrome). Serum calcitriol (1,25-dihydroxy vitamin D3, is the hormonally active metabolite of vitamin D) concentrations can be either low or inappropriately normal. In children, cystinosis, Wilson disease, and hereditary fructose intolerance are the most common of the syndrome.
· d) Drugs that can produce renal phosphate wasting include loop diuretics; acetazolamide (a carbonic anhydrase inhibitor diuretic); bisphosphonates (a class of drugs that prevent the loss of bone mass; used to treat osteoporosis and similar diseases), including pamidronate and zoledronate; and multiple chemotherapeutic and biologic agents, including cisplatinum; bevacizumab plus irinotecan, everolimus plus octreotide LAR; imatinib mesylate (a drug used in the treatment of chronic myelogenous leukemia and gastrointestinal stromal tumors); sorafenib; carmustine; and ifosfamide. In some cases, renal phosphate wasting is part of a more generalized, drug-induced Fanconi syndrome.
· e) Extracellular volume expansion or the administration of bicarbonate: can cause loss of phosphate through the kidneys.
· f) Oncogenic osteomalacia, a paraneoplastic syndrome characterized by osteomalacia, hypophosphatemia, renal phosphate wasting, bone pain, and muscle weakness. Several tumors that cause this syndrome have been described, most of which are benign tumors of mesenchymal origin.
· g) Other factors that can increase urinary phosphate excretion are: osmotic diuresis (most often due to glucosuria), proximally acting diuretics (acetazolamide and some thiazide diuretics that also have carbonic anhydrase inhibitory activity, such as metolazone), and acute volume expansion (which diminishes proximal sodium reabsorption).
· III) Intracellular shift of phosphate. Several physiologic agents stimulate phosphate uptake from the extracellular environment into the cell. This phenomenon can exacerbate the hypophosphatemia caused by the previously described mechanisms and can result in profound hypophosphatemia. However, in some circumstances, the shift alone may be enough to produce hypophosphatemia, albeit of a milder degree. Acute respiratory alkalosis or hyperventilation produces hypophosphatemia by stimulating a shift of phosphate into the cells; it is observed with salicylate overdose, panic attacks, and sepsis. Extreme hyperventilation in normal subjects can lower serum phosphate concentrations to below 1.0 mg/dL (0.32 mmol/L), and it is probably the most common cause of marked hypophosphatemia in hospitalized patients. Less pronounced hypophosphatemia may occur during the increase in ventilation after the successful treatment of severe asthma. The effects of respiratory alkalosis are exacerbated by concomitant glucose infusions and may persist after hyperventilation ceases. Respiratory alkalosis also may be the precipitating factor in the hypophosphatemia-induced acute rhabdomyolysis that can occur in alcoholic patients.
· Other mechanisms include:
· a) Treatment of diabetic ketoacidosis, refeeding, and parenteral nutrition therapy: insulin increases cell phosphate uptake and causes hypophosphatemia.
· b) Exogenous epinephrine also stimulates cellular phosphate uptake.
· c) Several cytokines reportedly stimulate intracellular phosphate shifts; this is perhaps responsible for the hypophosphatemia observed in the ICU setting of trauma, extensive burns, and bone marrow transplantation.
· d) In hungry bone syndrome, rapid uptake of phosphate into bone occurs after the initial treatment of osteomalacia or rickets or post-parathyroidectomy.
· e) Kidney transplantation. Hypophosphatemia is a common complication of kidney transplantation. Tertiary hyperparathyroidism has long been thought to be the etiology, but hypophosphatemia can occur in patients with low parathyroid hormone (PTH) levels and can persist after high PTH levels normalize. Moreover, even in the setting of normal allograft function, hypophosphatemia, and hyperparathyroidism, calcitriol levels remain inappropriately low following transplantation, suggesting that mechanisms other than PTH contribute to phosphate homeostasis. The factor FGF23 induces phosphaturia, inhibits calcitriol synthesis, and accumulates in chronic kidney disease, and has been suggested as a possible mediator of post-transplantation hypophosphatemia.
{kind=link}
· Hypophosphatemia diagnostic algorithm:
{kind=link}
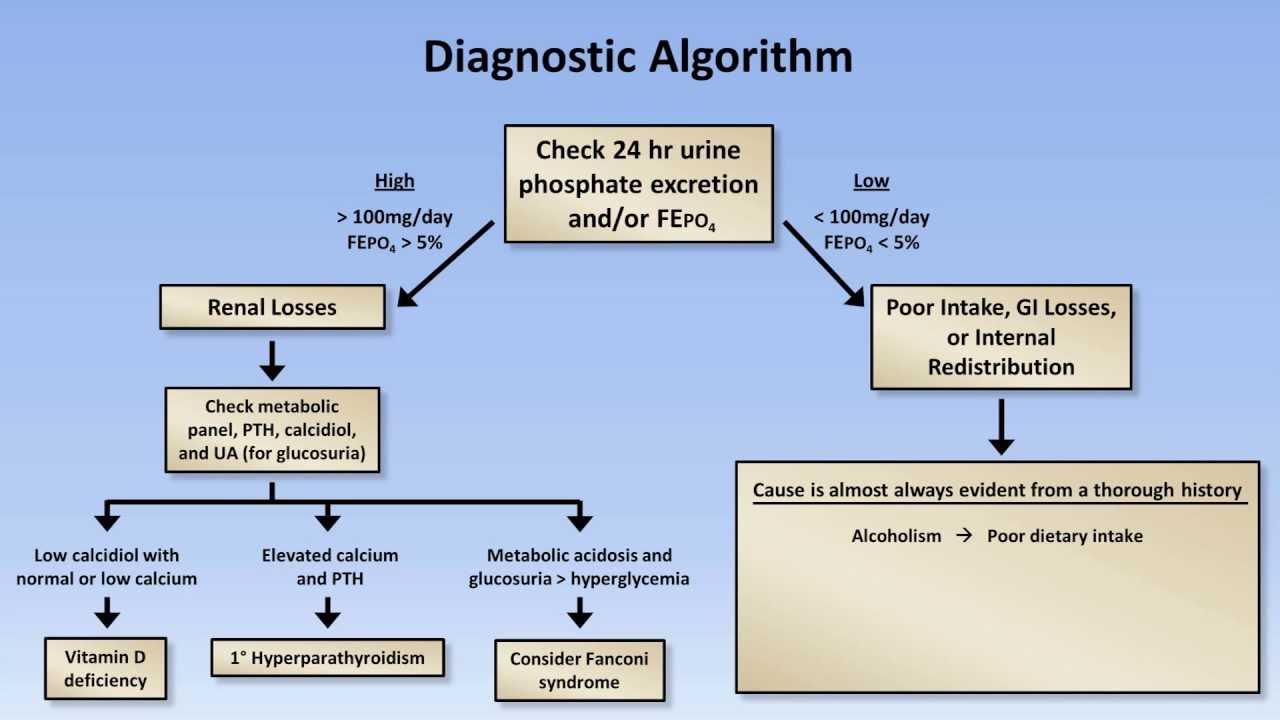{kind=link}
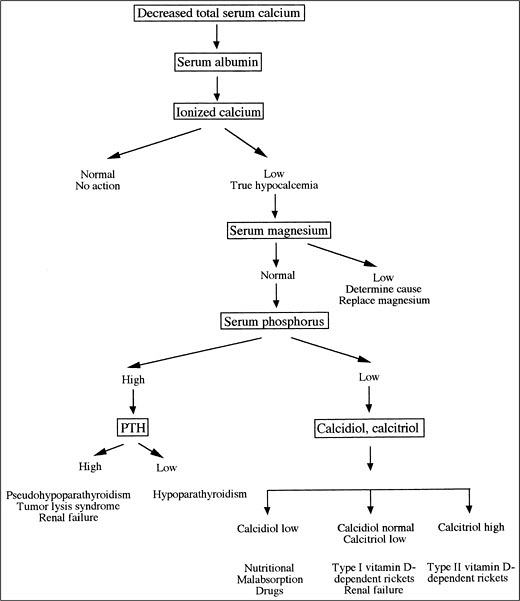{kind=link}
· Hyperphosphatemia: is abnormally high serum phosphate levels. They can result from increased phosphate intake, decreased phosphate excretion, or a disorder that shifts intracellular phosphate to extracellular space. Even severe hyperphosphatemia is for the most part clinically asymptomatic. Morbidity In patients with this condition is more commonly associated with an underlying disease than with increased phosphate values.
· Signs & symptoms: although most patients with hyperphosphatemia are asymptomatic, they occasionally report hypocalcemic symptoms, such as muscle cramps, tetany, and perioral numbness or tingling. Other symptoms include bone and joint pain, pruritus, and rash. More commonly, patients report symptoms related to the underlying cause of hyperphosphatemia. These generally are uremic symptoms, such as fatigue, shortness of breath (SOB), anorexia, nausea & vomiting, and sleep disturbances.
· In acute hyperphosphatemia, especially that caused by parenteral phosphate administration, the patient may be hypotensive or exhibit signs of hypocalcemia, such as positive Trousseau or Chvostek sign, hyperreflexia, carpopedal spasm, and seizures. Cataracts can be an ocular sign of hyperphosphatemia, but the cardiovascular (hypotension and heart failure, prolongation of the QT interval) and nervous systems are the most commonly affected by the condition. Central nervous system (CNS) and neuromuscular signs and symptoms in patients with hyperphosphatemia include altered mental status, delirium, obtundation, coma, convulsions and seizures, muscle cramps or tetany, neuromuscular hyperexcitability (i.e., Chvostek and Trousseau signs), and paresthesias (particularly perioral and distal extremities).
· Causes of hyperphosphatemia: the most common cause of hyperphosphatemia is renal failure. Less common causes can be classified according to pathogenesis. Often, several mechanisms contribute.
· Causes are related to:
· I) Increased intake. This can result from excessive oral or rectal use of an oral phosphate-saline laxative (Phospho-soda); excessive parenteral administration of phosphate; milk-alkali syndrome (*); and vitamin D intoxication.
· II) Decreased excretion. This can result from renal failure (acute or chronic); hypoparathyroidism; pseudohypoparathyroidism; severe hypomagnesemia; tumoral calcinosis; and bisphosphonate therapy.
· III) Shift of phosphate from intracellular to extracellular space. This can result from rhabdomyolysis; tumor lysis; acute hemolysis; and acute metabolic or respiratory acidosis.
· IV) Spurious. Results that falsely indicate the presence of hyperphosphatemia can result from a blood sample taken from a line containing heparin or alteplase; high concentrations of paraproteins; hyperbilirubinemia; in vitro hemolysis and hyperlipidemia.
· (*) Milk-alkali syndrome is characterized by high blood calcium caused by taking in too much calcium and absorbable alkali; common sources of calcium and alkali are dietary supplements taken to prevent osteoporosis and antacids. If untreated, milk-alkali syndrome may lead to kidney failure.
{kind=link}
{kind=link}
(ESRD: end-stage renal disease)
· Hyperphosphatemia diagnostic algorithm:
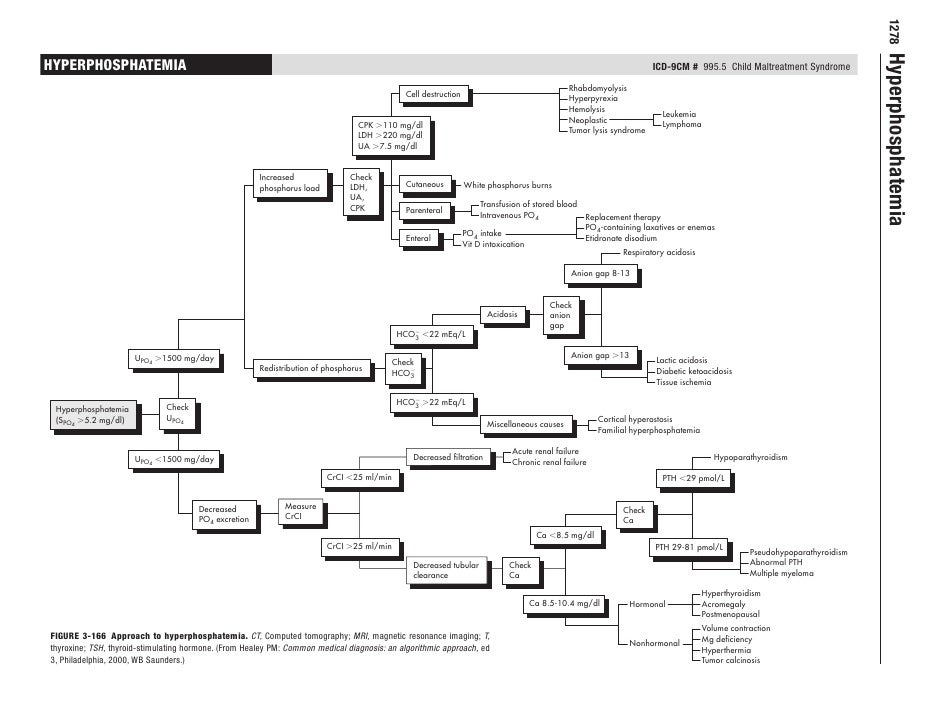{kind=link}
· Fractional excretion of phosphate (FEPi): it is measured with the formula
FEphosphate (FEPi)%= (Urine phosphate x serum Creatinine) / (serum phosphate x Urine Creatinine) x100
FEphosphate (FEPi)%= (Urine phosphate x serum Creatinine) / (serum phosphate x Urine Creatinine) x100
· FEPi < 10% (< 0.1 fraction) is low.
· FEPi > 20% (>0.2 fraction) is high.
· Tubular reabsorption of phosphate (%) = 100 – fractional excretion of phosphate.
· Tubular reabsorption of phosphate depends on plasma phosphate and glomerular filtration rate and is not a satisfactory indicator of tubular phosphate handling. This has led to the increasing use of ‘tubular maximum for phosphate corrected for GFR (TmP/GFR),’ a factor independent of plasma phosphate and renal functions for assessment of renal phosphate handling. TmP/GFR (normal 2.8 – 4.4 mg/dL) is an index of renal threshold for phosphate which can be determined by a nomogram4 (on timed urine samples) or directly as follows: TmP/GFR (mg/dL) = plasma phosphate — [(urine phosphate x plasma creatinine) / urine creatinine].
· In patients with hypophosphatemia a 4-hour urine phosphate excretion < 100 mg or fractional excretion of filtered phosphate (FEPO4) < 5% indicates appropriate low renal phosphate excretion, suggesting that the hypophosphatemia is caused by internal redistribution (e.g., refeeding syndrome, acute respiratory alkalosis) or decreased intestinal absorption (e.g., chronic antacid therapy, steatorrhea/ malabsorption).
· Fractional excretion of phosphate (FEPi) calculator:
SERUM & URINE OSMOLALITY
· Serum & Urine osmolality: osmolality and osmolarity are measurements of the solute concentration of a solution. In practice, there is a negligible difference between the absolute values of the different measurements. For this reason, both terms are often used interchangeably, even though they refer to different units of measurement.
· Osmolality is an estimation of the osmolar concentration of plasma and is proportional to the number of particles per kilogram of solvent. It is expressed as mOsmol/kg (the SI unit is mmol/kg, but mOsmol/kg is still widely used). Osmolality is measured by clinical laboratories using an osmometer (either a freezing point depression osmometer or a vapor pressure depression osmometer). The normal osmolality of extracellular fluid is 280 – k295 mOsmol/kg.
· Osmolarity is an estimation of the osmolar concentration of plasma and is proportional to the number of particles per liter of solution; it is expressed as mmol/l. This is what is used when a calculated value is derived. It is derived from the measured sodium (Na+), potassium (K+), urea, and glucose concentrations. The osmolarity is unreliable in various conditions, e.g., pseudo hyponatraemia such as hyperlipidemia in nephrotic syndrome, or hyperproteinemia.
· The following equations can be used to calculate serum osmolality:
Serum Osmolality Calculation (ethanol not always included) = 2 x (Na+) + (Glucose/18) + (BUN/2.8) + (Ethanol/3.8)
Serum Osmolality Calculation (ethanol not always included) = 2 x (Na+) + (Glucose/18) + (BUN/2.8) + (Ethanol/3.8)
· Note: Glucose, BUN, and ethanol may be reported in mg/dL (milligrams per deciliter) or mmol/L (millimole per liter). The numbers shown in the equation above are used to convert from mg/dL to mmol/L. For mmol/L, the equation would be: 2 x (Na+) + (Glucose) + (BUN) + (Ethanol)
· The term osmolarity has largely been superseded by osmolality, even when discussing calculated values.
· The osmotic gap (also called osmolar gap) is an arbitrary measure of the difference between the actual osmolality (measured by the laboratory) and the calculated osmolality.
Serum Osmotic Gap = Serum osmolality (measured) – serum osmolality (calculated).
Serum Osmotic Gap = Serum osmolality (measured) – serum osmolality (calculated).
· It is normally less than 10 – 15 mOsmol/Kg (range may differ between laboratories). Where the osmotic gap is increased, it indicates the presence of other osmotically active solutes which are not taken into account in the calculated osmolality, e.g., in methanol or ethylene glycol ingestion.
· A serum osmolality test and the osmotic gap may be ordered when a person has symptoms that a health practitioner suspects may be due to hyponatremia, such as polydipsia (excessive thirst), confusion, nausea, headache, lethargy, and in severe cases, seizures or coma. These tests may also be ordered when it is suspected that someone has ingested a toxin such as methanol or ethylene glycol.
· Urine osmolality is frequently ordered along with serum osmolality. It is used to help evaluate the body's water balance and to investigate increased and decreased urine output.
· Increased urine output may be due to increased fluid intake, lack of appropriate amounts of antidiuretic hormone (ADH), or diabetes where increased glucose levels lead to increased urine output.
· Decreased urine output may be due to a variety of causes, including decreased blood flow to the kidneys, an appropriate response to dehydration, or damage to the tubular cells in the kidneys.
· Urine sodium and creatinine are often ordered along with urine osmolality.
· Sometimes a urine osmotic gap is calculated and used to help evaluate the kidney's ability to excrete acid and reabsorb bicarbonate, to detect the presence of osmotically active molecules, and to compare with the serum osmotic gap. A urine osmolality test may be ordered along with blood testing when a health practitioner wants to compare urine results with the serum osmolality and/or when the person being tested is producing increased or decreased quantities of urine. Both may be ordered when it is suspected that the person may have diabetes.
· Osmolality testing may be ordered for asymptomatic people when unexplained hyponatremia is discovered during testing for other reasons.
· Serum and urine osmolality testing may be ordered frequently to monitor the effectiveness of treatment for these conditions and at regular intervals to monitor those taking the drug mannitol. As cell membranes, in general, are freely permeable to water, the osmolality of the extracellular fluid (ECF) is approximately equal to that of the intracellular fluid (ICF). Therefore, plasma osmolality is a guide to intracellular osmolality. Changes in ECF osmolality have a great effect on ICF osmolality and can cause problems with normal cell functioning and volume.
· In healthy people, increased osmolality in the blood will stimulate the secretion of antidiuretic hormone (ADH). This will result in increased water reabsorption, more concentrated urine, and less concentrated blood plasma.
· Diabetes insipidus is a condition caused by hyposecretion of, or insensitivity to, the effects of ADH. Low serum osmolality will suppress the release of ADH, resulting in decreased water reabsorption and more concentrated plasma. An increase of only 2% to 3% in plasma osmolality will produce a strong desire to drink. A change of 10% to 15% in blood volume and arterial pressure is required to produce the same response.
· Note: Stool osmolality may help evaluate chronic diarrhea that does not appear to be due to a bacterial or parasitical infection, i.,e. Feces may contain osmotically active substances (e.g., a laxative). The stool osmotic gap may also be calculated.
· Interpretation of serum & urine osmolality levels: In general, if someone has an increased serum osmolality, it is due to either decreased water in the blood or increased solutes. If the person has decreased serum osmolality, it is due to increased fluid levels.
· a) Normal or increased serum osmolality & increased urine osmolality: Causes include dehydration, renal disease & uremia, congestive heart failure (CHF), Addison’s disease, hypercalcemia, diabetes mellitus/ hyperglycemia, hypernatremia, alcohol ingestion, and mannitol therapy.
· b) Normal or increased serum osmolality & decreased urine osmolality: Causes include diabetes insipidus.
· c) Decreased serum osmolality & increased urine osmolality: Causes include syndrome of inappropriate ADH (antidiuretic hormone) secretion (SIADH).
· d) Decreased serum osmolality & decreased urine osmolality (with no increase in fluid intake: Causes include overhydration, hyponatremia, adrenocortical insufficiency, and sodium loss (diuretic or a low salt diet).
· Causes of increased serum osmolality: dehydration, sepsis, fever, sweating, burns; diabetes mellitus (hyperglycemia); diabetes insipidus; uremia; hypernatremia; mannitol therapy; ethanol, methanol or ethylene glycol or isopropyl alcohol ingestion/ poisoning; stroke, head trauma; shock; kidney damage.
· Increased urine output and a high osmolality may be seen when there is a substance being flushed from the body, such as excess glucose with diabetes mellitus.
· If a person has decreased urine output and high osmolality, then the person may be dehydrated.
· If someone has low or normal osmolality, the person may have kidney damage.
· Causes of increased urine osmolality: dehydration, congestive heart failure (CHF), syndrome of inappropriate ADH (antidiuretic hormone secretion (SIADH), adrenal insufficiency/ Addison’s disease, shock, liver damage, glycosuria, hypernatremia, high protein diet.
· Causes of decreased serum osmolality: excess hydration, hyponatremia, syndrome of inappropriate ADH (antidiuretic hormone) secretion (SIADH).
· When a person has increased urine output and low osmolality, then the person either is ridding their body of excess fluids or is unable to concentrate urine appropriately.
· Causes of decreased urine osmolality: diabetes insipidus, excess fluid intake, hypercalcemia, hypokalemia, kidney tubular damage, acute renal insufficiency, and glomerulonephritis.
· Οsmotic gap interpretation: When someone has an increased osmotic gap and is suspected of ingesting a toxin such as methanol, then it is likely that the person has done so, with the size of the gap related to the amount of toxin. During monitoring, if osmolality, the osmotic gap, and findings such as a low sodium level return to normal, then treatment has been effective.
· With mannitol therapy, the monitoring goal is the maintenance of a stable "therapeutic" osmotic gap and the relief of cerebral edema (swelling).
· Stool osmolality may sometimes be ordered to help evaluate chronic diarrhea that does not appear to be due to a bacterial or parasitic infection or to another identifiable cause such as intestinal inflammation or damage. People with chronic watery diarrhea may have an osmotically active substance, such as a commercial laxative, that is inhibiting the reabsorption of water by the intestines. Sometimes a stool osmotic gap is calculated. Stool osmolality may be ordered when a doctor suspects that a person's chronic diarrhea may be due to an osmotically active substance. If someone has an increased stool osmolality gap, then it is likely that an osmotically active element is causing their chronic liquid diarrhea. This can be seen with malabsorption and laxative abuse.
· Serum osmolality calculator:
· Urine osmolality calculator:
· Serum osmolality gap calculator:
· Urine osmolal gap calculator:
· Stool osmotic gap calculator:
No comments:
Post a Comment