
Dr. James Manos (MD)
January 2, 2016
Tips in Hematology
Volume (7)
2nd edition (revised)
CONTENTS
THROMBOCYTOPENIA
Thrombocytopenia
Causes of thrombocytopenia
Pseudothrombocytopenia
Wiskott–Aldrich syndrome (WAS)
Neonatal thrombocytopenia
Ιdiopathic thrombocytopenic purpura (ITP; also known as autoimmune thrombocytopenic purpura)
Post-transfusion purpura (PTP)
Thrombotic thrombocytopenic purpura (TTP; also known as Moschcowitz syndrome)
Hemolytic uremic syndrome (HUS)
Evans syndrome
DIC (disseminated intravascular coagulation)
Heparin-induced thrombocytopenia (HIT)
DISORDERS OF PLATELET FUNCTION
Disorders of platelet function
Bernard–Soulier syndrome (BSS)
Glanzmann's thrombasthenia
Platelet storage pool deficiency
Hermansky–Pudlak syndrome (HPS)
Chediak–Higashi syndrome
HEREDITARY COAGULATION ABNORMALITIES
Von Willebrand’s disease (vWD)
Haemophilia A
Haemophilia B
Acquired Haemophilia
THROMBOCYTOPENIA
· Thrombocytopenia: a disorder in which there is a relative decrease in platelets (thrombocytes) in peripheral blood. A normal human platelet count ranges from 150,000 to 450,000 platelets per microlitre of blood. These limits are determined by the 2.5th lower and upper percentile, so values outside this range do not necessarily indicate disease.
· One common definition of thrombocytopenia that requires emergency treatment is a platelet count below 50,000/ microlitre.
· Clinically – signs & symptoms: some patients may experience external bleeding such as epistaxis (nosebleeds) and/ or bleeding gums. Some women may have heavier or longer periods or breakthrough bleeding.
· Purpura (e.g., in the forearms) may also occur by spontaneous bleeding under the skin.
· Petechia (pinpoint hemorrhages in the skin and mucous membranes) may occur, e.g., on feet and legs.
· If the platelet count is between 30 000 and 50 000/mm3, bruising with minor trauma may be expected; spontaneous bruising will be seen if it is between 15 000 and 30 000/mm3.
· Causes of thrombocytopenia: decreased production of platelets (infections, megaloblastic anemia, aplastic anemia, drugs, bone marrow infiltration) or increased destruction of platelets [infections (e.g., HIV, malaria, dengue), DIC (disseminated intravascular coagulation), drugs [e.g., heparin, quinidine (an antiarrhythmic), gold salts], ITP (idiopathic thrombocytopenic purpura)].
· Specifically, the causes are classified into 3 categories:
· a) Decreased production: dehydration; vitamin B12 or folate (folic acid) deficiency; leukemia; myelodysplastic syndrome (MDS); aplastic anemia; liver failure (decreased production of thrombopoietin by the liver); sepsis, systemic viral or bacterial infection; dengue fever.
· b) Hereditary syndromes: congenital amegakaryocytic thrombocytopenia; thrombocytopenia-absent radius syndrome; Fanconi anemia; Bernard – Soulier syndrome (large platelets); May – Hegglin anomaly (thrombocytopenia, pale-blue leukocyte inclusions, and giant platelets); Grey platelet syndrome; Alport syndrome; Wiskott – Aldrich syndrome.
· c) Increased destruction: idiopathic thrombocytopenic purpura (ITP), thrombotic thrombocytopenic purpura (TTP); Hemolytic – uremic syndrome (HUS); disseminated intravascular coagulation (DIC); paroxysmal nocturnal hemoglobinuria, antiphospholipid syndrome; systemic lupus erythematosus (SLE); post-transfusion purpura; neonatal alloimmune thrombocytopenia; hypersplenism (splenic sequestration of platelets); dengue fever; Gaucher’s disease; and HIV – associated thrombocytopenia.
· d) Medication-induced:
· i) Direct myelosuppression: valproic acid (an antiepileptic); methotrexate (MTX); carboplatin, interferon; isotretinoin (for cystic acne, some types of ichthyosis and some type of cancer such as neuroblastoma); panobinostat (cancer chemotherapy); other chemotherapy medications; montelukast (for asthma and seasonal allergies), H2 blockers & PPIs (proton pump inhibitors) (the last 2 decreases gastric acid production and are used in peptic ulcer disease, oesophagitis, gastritis, gastroesophageal reflux disease, Zollinger – Ellison syndrome, etc.).
· ii) Immunological platelet destruction: heparin (heparin-induced thrombocytopenia (HIT); abciximab (abciximab – induced thrombocytopenia; abciximab is a glycoprotein IIb/IIIa receptor antagonist that is used in angioplasty in coronary heart disease).
· e) Other causes: snakebites (e.g., pit vipers); Onyalai (a disease of unknown etiology seen only in parts of Africa but suspected of being caused by poor nutrition or consumption of tainted food); excess consumption of oils containing erucic acid (e.g. Lorenzo’s oil or mustard oil); niacin toxicity (mainly when large doses are prescribed in patients with impaired renal function); Lyme disease (caused by the bacteria Borrelia; such as Borrelia burgdorferi sensu stricto in North America; transmitted by the ticks of the genus Ixodes); and pseudo-thrombocytopenia.
· For extended causes, see also:
· http://www.aafp.org/afp/2012/0315/p612.html (table 2)
· Pseudothrombocytopenia: is a relatively uncommon phenomenon caused by in vitro agglutination of platelets. As a result of platelet clumping, platelet counts reported by automated analyzers may be much lower than the actual number in the blood because these devices cannot differentiate platelet clumps from individual cells. The incidence of pseudo-thrombocytopenia reported in different studies ranges from 0.09 to 0.21%, which accounts for 15 to 30% of all cases of isolated thrombocytopenia. It has been associated with the use of ethylenediaminetetraacetic acid (EDTA) as an anticoagulant, platelet cold agglutinins, and multiple myeloma.
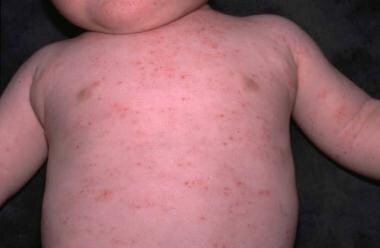
· Wiskott–Aldrich syndrome (WAS; also called eczema – thrombocytopenia –immunodeficiency syndrome): a rare Χ – linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea (due to low platelets). Due to its mode of inheritance, most patients are male. The first signs of WAS are usually petechiae and bruising resulting from a low platelet count. Spontaneous epistaxis and bloody diarrhea are common.
· Eczema develops within the first month of life, and recurrent bacterial infections develop for three months.
· Splenomegaly is not uncommon.
· The majority of WAS children develop at least one autoimmune disorder, and cancers (leukemia and lymphoma) develop in up to one-third of patients.
· Lab features – immunological studies: immunoglobulin M (IgM) levels are reduced, IgA and IgE are elevated, and IgG levels can be normal, reduced, or elevated.
{kind=link}
{kind=link}
{kind=link}
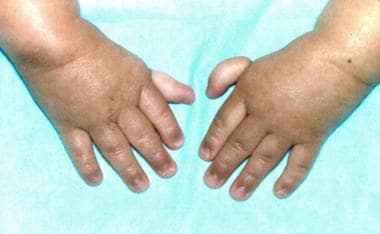
· Fanconi anemia (FA): (it should not be confused with Fanconi syndrome, a kidney disorder) Fanconi anemia is a genetic disease with an incidence of 1: 350,000 births, with a higher frequency in Ashkenazi Jews (the carrier frequency in the Ashkenazi Jewish population is about 1/90) and Afrikaners in South Africa.
· It is primarily an autosomal recessive genetic disorder. There is a 25% risk that each subsequent child will have FA. About 2% of FA cases are X-linked recessive, which means that if the mother carries one mutated Fanconi anemia allele on one X chromosome, there is a 50% chance that male offspring will present with Fanconi anemia. Scientists have identified 17 FA or FA-like genes.
· FA is the result of a genetic defect in a cluster of proteins responsible for DNA repair. As a result, most FA patients develop cancer, most often acute myelogenous leukemia (AML), and 90% develop bone marrow failure by age 40. About 60–75% of FA patients have congenital defects, commonly short stature, abnormalities of the skin, arms (such as radial deficiencies), head, eyes, kidneys, and ears, and developmental disabilities.
· About 75% of FA patients have some form of endocrine problem, with varying degrees of severity. The median age of death was 30 years in 2000. Short stature and skin pigmentation may become apparent during childhood, including cafe au lait spots. The first sign of a hematologic problem is usually petechiae and bruises, with a later onset of paleness & tiredness (anemia) and infections.
· Because macrocytosis usually precedes thrombocytopenia, patients with typical congenital anomalies associated with FA should be evaluated for an elevated MCV.
· Treatment with androgens and hematopoietic (blood cell) growth factors can help bone marrow failure temporarily, but the long-term treatment is a bone marrow transplant. Cells from people with FA are sensitive to drugs that treat cancer by DNA crosslinking, e.g., mitomycin C.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
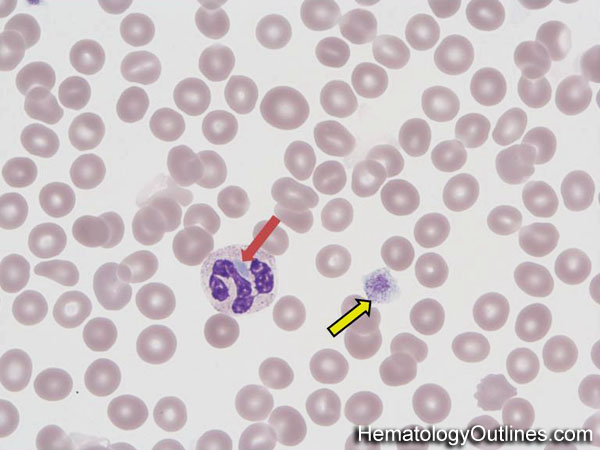
· Gray platelet syndrome (GPS): Gray platelet syndrome, also called platelet alpha-granule deficiency, is a rare congenital autosomal recessive bleeding disorder caused by a reduction or absence of alpha–granules in platelets and the release of proteins normally contained in these granules into the marrow, causing myelofibrosis. GPS is primarily inherited in an autosomal recessive manner (NBEAL2 gene at chromosome 3p). GPS is characterized by thrombocytopenia and abnormally large agranular platelets in peripheral blood smears. Patients with the GPS have mild to moderate bleeding tendencies.
· Alport syndrome: Alport syndrome, also called hereditary nephritis, is a genetic disorder characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also cause eye abnormalities, including cataracts, lenticonus, keratoconus, and retinal flecks in the macula & mid-periphery. Urine may be positive for blood and protein. Platelet disorders include thrombocytopenia or granulocytic inclusions, like the May – Hegglin anomaly (see below).
· May – Hegglin anomaly: a rare genetic disorder characterized by thrombocytopenia, pale-blue leukocyte inclusions (Dohle–like bodies), and giant platelets.
{kind=link}
{kind=link}
{kind=link}
· Neonatal thrombocytopenia: Most cases of thrombocytopenia affect preterm birth infants and result from placental insufficiency and/or fetal hypoxia. The other causes are less frequent, e.g., alloimmune, genetic, autoimmune, infection, and DIC. Thrombocytopenia that starts after the first 72 hours since birth is often the result of underlying sepsis or necrotizing enterocolitis (NEC).
· In the case of infection, possible pathogens may be fungi, bacteria, and viruses, for example, CMV, rubella, HIV, Streptococcus agalactiae, Listeria monocytogenes, E.coli, Haemophilus influenza, toxoplasmosis, Yersinia enterocolitica, etc.
· Ιdiopathic thrombocytopenic purpura (ITP; also known as autoimmune thrombocytopenic purpura, immune thrombocytopenia, primary immune thrombocytopenia, and primary immune thrombocytopenic purpura): is defined as isolated thrombocytopenia with normal bone marrow and the absence of other causes of thrombocytopenia.
· It causes a characteristic purpuric rash and an increased tendency to bleed.
· Spontaneous bleeding may occur when the platelet count is less than 20,000/mm3
· Demographics: In chronic ITP (adults), the female-to-male ratio is 2.6:1. More than 72% of patients older than 10 years are female. In acute ITP (children), the distribution is equal between males (52%) and females (48%). In adults, the peak prevalence is from 20 – 50 years of age. In children, the peak prevalence is from 2 – 4 years of age. About 40% of all patients are younger than 10 years.
· In children, ITP is more common in boys than in girls. In middle-aged adults, women are affected more frequently than men. Children may develop ITP at any age, but the incidence peaks in children aged 1 – 6 years. Adults may be affected at any age, but most cases are diagnosed in women aged 30-40 years.
· Onset in a patient older than 60 years is uncommon, and a search for other causes of thrombocytopenia is warranted. The most likely causes in these persons are myelodysplastic syndromes (MDS), acute leukemia, and marrow infiltration (myelophthisis). Persons with ITP who are 70 years or older are at increased risk for spontaneous bleeding and treatment-related adverse events.
· Pathophysiology: ITP is an autoimmune disease with IgG antibodies detectable against several platelet surface antigens. There is immune destruction of platelets due to the reaction of antibodies with platelet antigens (such as Gp IIb/ IIIa).
· Two distinct clinical syndromes manifest as an acute condition in children and a chronic condition in adults.
· In children, most immune thrombocytopenic purpura (ITP) cases are acute, manifesting a few weeks after a viral illness. In adults, most cases of ITP are chronic, manifesting with an insidious onset, and occur in middle-aged women.
· Pregnant women with immune thrombocytopenic purpura (ITP) can be asymptomatic or can present with a history of easy bruising, bleeding into the mucous membranes (epistaxis or gingival bleeding), or petechiae. They may have a history of menorrhagia or menometrorrhagia prior to pregnancy. Maternal history of delivering a term newborn with thrombocytopenia, visceral or intracranial hemorrhage, or spontaneous or prolonged bleeding after venipuncture or circumcision raises suspicion for neonatal alloimmune thrombocytopenia (NAIT). However, about 50% of neonates with NAIT are first-born children and thus are delivered to women whose risk for the disorder is previously unrecognized and unknown.
· Acute ITP often follows an infection (e.g., a viral infection). Thrombocytopenia is severe, but the disease is self-limiting and has a spontaneous resolution within 2 months.
· Chronic ITP occurs in young adults with an insidious onset of bleeding and with an unknown cause. Thrombocytopenia is moderate. The disease persists longer than 6 months and may last for many years with exacerbations and remissions.
· Clinically features are purpura, epistaxis, menorrhagia, or bleeding from gums.
· Causes: The diagnosis of ITP is a process of exclusion. First, it has to be determined that there are no blood abnormalities other than a low platelet count and no physical signs other than bleeding. Then, secondary causes (5–10% of ITP cases) should be excluded. Secondary causes include leukemia, medications (e.g., heparin, quinine), antiphospholipid syndrome, SLE (systemic lupus erythematosus), HIV, hepatitis C, liver cirrhosis, congenital causes, von Willebrand factor deficiency, onyalai (a type of acquired immune thrombocytopenia limited in back populations in central southern Africa; the characteristic manifestation is hemorrhagic bullae on the mucosa of the oral-nasopharynx), etc. In about 1% of ITP cases, autoimmune hemolytic anemia and ITP coexist. This is called Evans syndrome, and CLL (chronic lymphocytic leukemia) should be excluded.
· In some people, ITP may be linked to viral or bacterial infections, such as HIV, hepatitis C, or H. pylori.
· Children who have acute (short-term) ITP often have had recent viral infections. These infections may "trigger" or set off the immune reaction that leads to ITP.
· While some cases of ITP are caused by drugs and others are associated with infection, pregnancy, or immune disorders such as systemic lupus erythematosus (SLE), about half of all cases are classified as "idiopathic," meaning the cause is unknown.
· Despite the destruction of platelets by splenic macrophages, the spleen is normally not enlarged, so an enlarged spleen should lead to a search for other possible causes for thrombocytopenia. Bleeding time is usually prolonged in ITP patients. However, a normal bleeding time does not exclude platelet disorders.
· Diagnosis: ITP is diagnosed by a low platelet count in a CBC (complete blood count). However, since the diagnosis depends on the exclusion of other causes of a low platelet count, additional investigations (such as a bone marrow biopsy) may be necessary in some cases.
· Blood smear shows a reduction in the number of platelets and occasional large–sized platelets.
· Bone marrow examination may be performed on patients over the age of 60 and those who do not respond to treatment or when the diagnosis is in doubt. The bone marrow biopsy may show an increase in the production of megakaryocytes.
· Assays for platelet antigen-specific antibodies, platelet-associated immunoglobulin, or other antiplatelet antibodies are available in some medical centers. The reliability of the results of a platelet antibody test is highly specific to the laboratory used. A negative antiplatelet antibody assay result does not exclude the diagnosis of ITP.
· Specific tests: There are two excellent tests to measure platelet-associated and plasma autoantibody: the immunobead assay and the monoclonal antibody-specific immobilization of platelet antigens (MAIPA) assay. Both tests measure autoantibody against antigens on specific platelet glycoprotein complexes (a complex is a combination of two or more proteins), usually glycoprotein IIb/IIIa (a combination of platelet glycoprotein IIb and platelet glycoprotein IIIa) and glycoprotein Ib/IX (a combination of platelet glycoprotein Ib and platelet glycoprotein IX). These tests give positive results in about 60% of ITP patients and are not positive in patients with other causes of thrombocytopenia. Unfortunately, these specific tests are done primarily in research laboratories and are unavailable in most clinical laboratories.
· Non-specific test: The test used in most clinical laboratories measures platelet-associated IgG. This detects any IgG antibody (both specific antiplatelet antibody and any other IgG antibody associated with the platelet). Although this test gives positive results in about 90% of chronic ITP patients, it may also be falsely positive in patients who have other causes of a low platelet count that are not due to antiplatelet autoantibody. Therefore, this test does not specifically measure the antiplatelet antibody and most experts do not recommend it.
· The direct assay for the measurement of platelet-bound autoantibodies has an estimated sensitivity of 49 – 66% and an estimated specificity of 78 – 92%. A negative test does not exclude the diagnosis. Many women with gestational thrombocytopenia have elevated levels of circulating platelet-associated immunoglobulin. Therefore, current antiplatelet antibody assays cannot be used to differentiate between ITP and gestational thrombocytopenia.
· Positive anti-platelet antibodies have 80% specificity for the diagnosis.
· About direct estimation with flow cytometry, a study concluded that a positive result indicates the presence of antibodies bound to the platelet surface. Positive results are observed in patients with idiopathic thrombocytopenic purpura (ITP), systemic lupus erythematosus (SLE), lymphoma, HIV infection, and drug-induced thrombocytopenia after quinidine, quinine, sulfonamides, gold salts, heparin, and other drug therapy. Positive results may also be seen in neonatal alloimmune thrombocytopenia and post-transfusion purpura; however, the indirect platelet antibody test is recommended when evaluating these 2 disorders since it is more likely to yield positive results. The platelet glycoprotein antibody ELISA method should confirm all positive results for suspected immune thrombocytopenia. A negative result suggests a non-immune etiology in patients with thrombocytopenia. Results from this test should be interpreted in context with all clinical and laboratory findings (Reference (Retrieved (20 December 2015)): http://www.questdiagnostics.com/testcenter/testguide.action?dc=TH_PlateletAb_Direct )
· Differential diagnosis: DIC, HIV, TTP.
· Treatment: in mild cases, only careful observation may be required, but very low counts or significant bleeding may prompt treatment with corticosteroids, IV immunoglobulin, anti–D immunoglobulin, or immunosuppressive drugs. Refractory ITP may require splenectomy. Platelet transfusions may be used in severe bleeding together with a very low platelet count. Sometimes, the body may compensate by making abnormally large platelets.
· Drug-induced thrombocytopenia:
· Drug-induced thrombocytopenia, also called drug-induced immune thrombocytopenia (DITP), is frequently associated with drug-induced antibodies that cause platelet destruction or clearance by the reticuloendothelial system (RES). Immune-mediated thrombocytopenia may also be caused by other substances, such as herbal remedies, foods, and beverages. All these causes are described by the term DITP. Less common mechanisms of drug-induced thrombocytopenia include bone marrow suppression by drugs other than known cytotoxic (e.g., chemotherapy) agents (often dose-dependent), and immune thrombocytopenia (ITP)-like syndrome in which autoimmune platelet destruction continues in the absence of the implicated agent. In most cases, thrombocytopenia is the only hematologic manifestation of drug toxicity.
· However, there are exceptions to this general rule:
· a) Heparin-induced thrombocytopenia (HIT) is associated with a hypercoagulable state and thrombosis rather than bleeding. HIT is discussed separately.
· b) Some drug-induced disorders, such as aplastic anemia and thrombotic microangiopathy (DITMA), result in thrombocytopenia along with other cytopenias and organ involvement.
· Commonly implicated drugs and their mechanisms include the following: abciximab (a glycoprotein IIb/IIIa receptor antagonist drug used for angioplasty; mechanism: antibody-mediated), beta-lactam antibiotics (e.g., penicillins, cephalosporins; mechanism: antibody-mediated), carbamazepine (antibody-mediated), eptifibatide & tirofiban (glycoprotein IIb/IIIa receptor antagonist drugs used for angioplasty; mechanism: antibody-mediated), gold compounds (bone marrow suppression), heparin (also antibody-mediated thrombosis), linezolid (antibiotic; mechanism: bone marrow suppression), MMR (measles mumps rubella) vaccine (ITP-like syndrome), phenytoin (antibody-mediated), piperacillin (antibiotic; mechanism: antibody-mediated), quinine & quinidine (antibody-mediated or thrombotic microangiopathy), rifampicin (antibody-mediated), sulfonamides & sulfamethoxazole-trimethoprim (SMX – TMP) (antibody-mediated), valproic acid (bone marrow suppression) and vancomycin (antibody-mediated).
· The thrombocytopenia is typically isolated (i.e., unaccompanied by anemia, leukopenia, leukocytosis, or coagulation abnormalities) and severe (i.e., platelet count <20,000/microL with clinical bleeding).
· Diagnosis: the diagnosis of drug-induced thrombocytopenia is made by documenting the prompt resolution of thrombocytopenia after discontinuation of the suspected drug (typically within one week). In some cases, drug-specific antiplatelet antibodies performed at specialized laboratories can confirm the diagnosis, but their absence does not exclude it.
· Differential diagnosis: the evaluation of a patient with suspected drug-induced thrombocytopenia differs between previously asymptomatic patients seen in an outpatient (office) setting, where DITP & ITP are more likely, and acutely ill hospitalized patients where sepsis & DIC are more often considerations. Congenital and/or acquired hematologic disorders associated with thrombocytopenia are suspected when there is another apparent cause (e.g., pregnancy, connective tissue disorders, infection) or when evaluation of the complete blood count (CBC/FBC) and peripheral smear shows the presence of abnormal platelet morphology (e.g., Bernard-Soulier syndrome), abnormal cellular maturation (e.g., MDS), pancytopenia (e.g., aplastic anemia), or the presence of abnormal circulating white blood cells (e.g., the acute and chronic leukemias).
· Post-transfusion purpura (PTP): is an adverse reaction to a blood or platelet transfusion that occurs when the body produces alloantigens to the introduced platelets’ antigens (antiplatelet antigens). These alloantibodies destroy the patient's platelets, leading to thrombocytopenia.
· PTP usually presents 5 – 12 days after transfusion and is a potentially fatal condition. PTP is rare but often occurs in women who have had multiple pregnancies or in men who have undergone previous transfusions.
· Pathophysiology: The precise mechanism leading to PTP is unknown, but it most commonly occurs after transfusion on HPA – 1a (-) recipient (individuals whose platelets lack the HPA (human platelet antigen) – 1a) or HPA – 1a (+) donor.
· The patient develops antibodies to the HPA-1a antigen, leading to platelet destruction.
In some cases, HPA-5b has also been implicated.
In some cases, HPA-5b has also been implicated.
· Treatment: It is usually self-limiting, but intravenous immunoglobulin (IVIG) therapy is the primary treatment.
· Plasmapheresis may also be used. It is essential to identify the donor to exclude the donor from further blood donations.
· Thrombotic thrombocytopenic purpura (TTP; also known as Moschcowitz syndrome): decreased Platelets, MHA (microangiopathic hemolytic anemia), fluctuating neurological signs, renal impairment, fever.
· MHA (microangiopathic hemolytic anemia): increased LDH, increased indirect (unconjugated) bilirubin, and schistocytes on a blood smear.
· Fluctuating neurological signs: e.g., hallucinations, altered mental status, headache, stroke, bizarre behavior, seizures, hemiplegia, aphasia, visual problems, paresthesia, TIA (transient ischemic attack), transient sensorimotor deficits, etc.
· Treatment: plasmapheresis (exchange transfusion), corticosteroids, rituximab (if resistant).
· Hemolytic uremic syndrome (HUS): hemolytic anemia, AKI (acute kidney injury), thrombocytopenia.
· Predominantly, but not exclusively, on children.
· Most cases are preceded by infectious diarrhea (may be bloody), e.g., by enterohemorrhagic E. coli (especially the Ο157: Η7 serotype), Campylobacter, Shigella, or viruses.
· Mortality 5 – 10%.
· A minority develops chronic kidney disease.
· Signs & symptoms: oliguria, hematuria, kidney failure, low platelets, hypertension.
· Microangiopathic hemolytic anemia (MHA): increased LDH, increased indirect (unconjugated) bilirubin, and schistocytes on a blood smear.
· Neurological signs may also occur in some cases.
· Treatment is supportive, e.g., dialysis on renal failure.
· Evans syndrome: an autoimmune disease where the individual’s Abs (antibodies) attack their own RBCs & platelets. Both events may co-occur, or one may follow on from the other. Other antibodies may occur directed against neutrophils and lymphocytes.
· Its overall pathology resembles a combination of autoimmune hemolytic anemia and idiopathic thrombocytopenic purpura.
· About 10 – 23% of patients who have autoimmune hemolytic anemia will also have thrombocytopenia and, thus, Evans syndrome.
· Diagnosis: blood tests to confirm hemolytic anemia, idiopathic thrombocytopenic purpura, a positive DAT (direct Coombs), and an absence of any known underlying cause.
· Treatment: initial treatment is with glucocorticoid corticosteroids or IV (intravenous) immunoglobulin. Relapses are not uncommon, and immunosuppressive medications may be used.
· Other treatment options include rituximab (in acute and refractory cases) and, in some instances, splenectomy. The only prospect for a permanent cure is the high-risk option of an allogenic hematopoietic stem cell transplantation (SCT).
· DIC (disseminated intravascular coagulation):
· DIC is described analytically above.
· Lab tests show reduced platelets, increased PT, aPTT, D-Dimers, and decreased fibrinogen (but, as it is an acute phase reactant, it may be normal or even increased by 57%).
· Mortality 10 – 50%.
· Many causes (trauma, sepsis, obstetric emergencies, etc.).
· Heparin-induced thrombocytopenia (HIT): the development of thrombocytopenia due to the administration of various forms of the anticoagulant heparin. The risk is greater with UFH (unfractionated heparin) than with LMWH (low molecular weight heparin). In orthopedic patients, the risk is 5% with UFH and 0.5% with LMWH.
· HIT predisposes to thrombosis; when thrombosis is identified, the condition is called heparin-induced thrombocytopenia and thrombosis (HITT).
· HIT is caused by the formation of abnormal antibodies that activate platelets.
· Clinical features: HIT is characterized by thrombocytopenia. However, it is generally not low enough to lead to an increased risk of bleeding. Most people with HIT will, therefore, not experience any symptoms. Typically, the platelet count will fall 5 – 14 days after heparin is first given. Severe thrombocytopenia < 15 X 109 is unusual. If someone has received heparin in the previous three months, the fall in platelet count may occur sooner, sometimes within a day.
· Also, 50% of patients have associated thrombosis. HIT may take the form of clots either in arteries or veins, causing arterial or venous thrombosis, respectively. Examples of arterial thrombosis are stroke, myocardial infarction (MI), and acute leg ischemia. Venous thrombosis may occur in the leg or arm in the form of deep vein thrombosis (DVT) or pulmonary embolism (PE).
· In those receiving heparin through an intravenous infusion, a complex of symptoms ("systemic reaction") may occur when the infusion is started. These include fever, chills, hypertension, tachyarrhythmia, dyspnea, and chest pain. This happens to about a quarter of people with HIT. Others may develop a skin rash consisting of petechiae.
· Diagnosis: HIT may be suspected if blood tests show a falling platelet count in someone receiving heparin, even if the heparin has already been discontinued. Professional guidelines recommend that people receiving heparin have a CBC (complete blood count) on a regular basis while receiving heparin. However, not all people with a falling platelet count while receiving heparin turn out to have HIT.
· The timing, severity of the thrombocytopenia, the occurrence of new thrombosis, and the presence of alternative explanations all determine the likelihood that HIT is present.
· A commonly used score to predict the likelihood of HIT is the "4 Ts" score which takes into account the following parameters:
· a) Thrombocytopenia
· b) Timing of platelet count fall
· c) Thrombosis or other sequelae
· d) Other causes of thrombocytopenia
· For a 4Ts score calculators see
· Also, someone suspected of having HIT antibodies against heparin-PF4 complexes may be detected with an ELIZA (enzyme-linked immunosorbent assay) test. However, the ELISA test detects all circulating antibodies that bind heparin-PF4 complexes and may also falsely identify antibodies that do not cause HIT. Therefore, those with a positive ELISA are tested further with a functional assay. This test uses platelets and serum from the patient; the platelets are washed and mixed with serum and heparin. The sample is then tested with a serotonin release assay (SRA) for the release of serotonin (a marker of platelet activation). If this shows high serotonin release, then the diagnosis of HIT is confirmed.
· If someone has been diagnosed with HIT, some recommend routine Doppler sonography of the leg veins to identify deep vein thromboses (DVT), as this is very common in HIT.
· Treatment: given the fact that HIT predisposes strongly to new episodes of thrombosis, it is not sufficient to simply discontinue the heparin administration. There is a risk of thrombosis if heparin is stopped. Alternative anticoagulation may be considered, such as direct thrombin inhibitor or danaparoid.
· The treatment of HIT requires both protection from thrombosis and the choice of an agent that will not reduce the platelet count further.
· 3 agents are used to provide anticoagulation in those with strongly suspected or proven HIT: danaparoid, lepirudin, and argatroban. These are alternatives to heparin therapy. There also have been case reports of fondaparinux being used in anti-coagulated patients with an established HIT as it has no affinity to PF-4. However, its renal excretion precludes its use in patients with renal dysfunction.
DISORDERS OF PLATELET FUNCTION
· Disorders of platelet function:
· a) Inherited (rare): Bernard – Soulier syndrome, Glanzmann’s thrombasthenia, storage pool deficiency (alpha granule & dense granule), defective thromboxane synthesis.
· b) Acquired (common): chronic myeloproliferative disorders, acute leukemia, MDS (myelodysplastic syndrome), paroxysmal nocturnal hemoglobinuria, paraproteinemias, uremia, cardiopulmonary bypass, drugs.
· Bernard–Soulier syndrome (BSS; also called hemorrhagiparous thrombocytic dystrophy): a rare autosomal recessive coagulopathy where there is a deficiency of glycoprotein GpIb receptor of platelet surface (the receptor for von Willebrand factor), leading to insufficient adhesion of platelets to the subendothelium.
· Incidence: less than 1 case per million persons. BSS is characterized by giant platelets.
· Clinical features: bleeding diathesis: gingival bleeding (bleeding gums), easy bruising, menorrhagia (heavy & prolonged menstrual periods), epistaxis, perioperative and postoperative bleeding, abnormally prolonged bleeding from small injuries.
· Glanzmann's thrombasthenia: an abnormality of the platelets; an extremely rare coagulopathy in which the platelets contain poor or low levels of glycoprotein GpII/IIIa, which is a receptor for fibrinogen. As a result, no fibrinogen bridging of platelets to other platelets can occur, and the bleeding time is significantly prolonged (PT, APTT, and platelet count are unaffected).
· Clinical features: bleeding diathesis: gingival bleeding (bleeding gums), easy bruising, menorrhagia (heavy & prolonged menstrual periods), epistaxis, perioperative and postoperative bleeding, abnormally prolonged bleeding from small injuries, postpartum bleeding, and gastrointestinal (GI) bleeding.
· Lab features: platelet aggregation is normal with ristocetin, but impaired with other agonists such as ADP, thrombin, collagen, or epinephrine.
· Platelet storage pool deficiency: a type of coagulopathy characterized by defects in the granules in platelets, notably a lack of granular non-metabolic ADP. Patients with ADP-deficient "Storage Pool Disease" present a prolonged bleeding time due to impaired aggregation response to fibrillar collagen.
· It may involve the alpha granules (gray platelet syndrome; Quebec platelet disorder) or the dense granules (delta–storage pool deficiency; Hermansky-Pudlak syndrome; Chediak–Higashi syndrome).
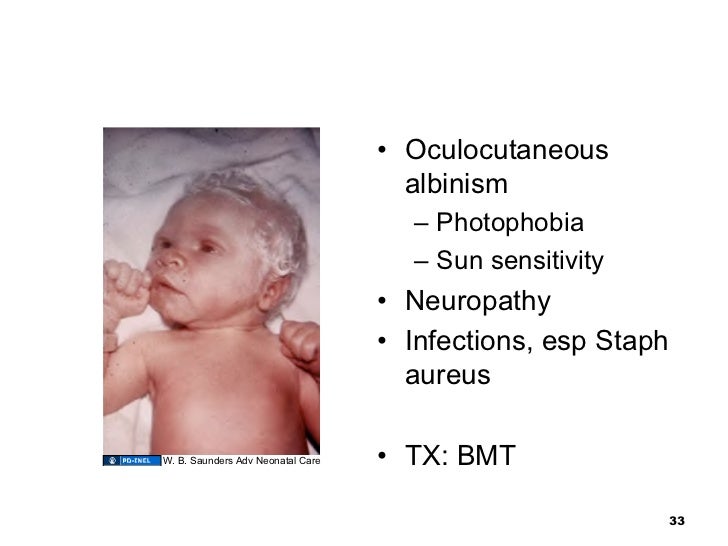
· Hermansky–Pudlak syndrome (HPS): a rare autosomal recessive disorder that results in occulo-cutaneous albinism, bleeding problems due to platelet storage pool defect, and lysosomal accumulation of ceroid lipofuscin (storage of an abnormal fat-protein compound).
· Incidence: it affects around 1 in 500 000 people worldwide, with a significantly higher occurrence in Puerto Rico (prevalence of 1 in 1 800).
· There are eight classic forms of the disorder based on the genetic mutation from which the disorder stems. A ninth type has also been described and is due to a mutation in the gene Pallidin.
· This syndrome may cause pulmonary fibrosis.
{kind=link}
· Chediak–Higashi syndrome: a rare autosomal recessive disorder that arises from a mutation of a lysosomal trafficking regulator protein, which leads to a decrease in phagocytosis that results in recurrent pyogenic infections, partial occulo-cutaneous albinism, and peripheral neuropathy.
· People with CHS have light skin and silvery hair (partial albinism) and frequently complain of solar sensitivity and photophobia.
· Neutropenia is common, leading to frequent infections, especially with Staphylococcus aureus and Streptococci. Also, neuropathy is common. The infections involve mucous membranes, skin, and the respiratory tract. Affected children are susceptible to infections.
· It is also associated with periodontal disease of deciduous dentition and bleeding disorders.
· The diagnosis is confirmed by bone marrow smears that show "giant inclusion bodies" in the cells that develop into white blood cells (leukocyte precursor cells). CHS can be diagnosed prenatally by examining a sample of hair from a fetal scalp biopsy or testing leukocytes from a fetal blood sample.
![http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-[4-bl050-4].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-%5b4-bl050-4%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
· http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-[3-bl050-3].jpeg?Width=600&Height=450&Format=4http://image.slidesharecdn.com/011209-jlevine-myeloidcelldisorders-110727140520-phpapp01/95/011209-myeloid-cell-disorders-33-728.jpg?cb=1311775763
![http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-[3-bl050-3].jpeg?Width=600&Height=450&Format=4](http://www.pathpedia.com/education/eatlas/histopathology/blood_cells/chediak-higashi_syndrome/chediak-higashi-syndrome-%5b3-bl050-3%5d.jpeg?Width=600&Height=450&Format=4){kind=link}
{kind=link}
HEREDITARY COAGULATION ABNORMALITIES
· Von Willebrand’s disease (vWD): defect in von Willebrand factor (vWF). The most common hereditary coagulation abnormality described in humans, although it can also be acquired as a result of other medical conditions. It arises from a qualitative or quantitive deficiency of the von Willebrand factor (vWF), a multimeric protein that is required for platelet adhesion.
· The prevalence of vWD is about 1 in 100 individuals. However, most of these people do not have symptoms. The prevalence of clinically significant cases is 1 per 10,000.
· Because most forms are rather mild, they are detected more often in women whose bleeding tendency shows during menstruation.
· VWD may be more severe or apparent in people with blood type O.
· There are three types of hereditary vWD: The four genetic types of vWD described are:
· a) Type 1
· b) Type 2
· c) Type 3
· d) Pseudo or platelet-type.
· There are various subtypes.
· Most cases are hereditary, but acquired forms of vWD have been described.
· Acquired vWD can occur in patients with autoantibodies (in this case the function of vWF is not inhibited, but the vWF-antibody complex is rapidly cleared from the circulation) or in patients with aortic stenosis (it may lead to gastrointestinal bleeding (Heyde’s syndrome)), or in patients with an implant of a Left Ventricular Assist Device (LVAD) (large multimers of vWF are destroyed by mechanical stress in both conditions).
· Also, it may be caused by thrombocythemia due to the sequestration of the von Willebrand factor via the adhesion of vast numbers of platelets. Acquired vWD has also been described in Wilm’s tumor (a cancer of the kidneys that usually affects children), hypothyroidism, and mesenchymal dysplasias.
· Clinical features: vWD Type I is the most common type of the disorder and those that have it are typically asymptomatic or may experience mild symptoms such as epistaxis (nosebleeds), although there may be severe symptoms in some cases.
· The distinct types of vWD present with varying degrees of bleeding tendency, usually in the form of easy bruising, epistaxis, and bleeding gums. Women may experience menorrhagia and blood loss during childbirth. Severe internal or joint bleeding is uncommon (except for in vWD type III).
· Diagnosis: measuring the amount of vWF in a vWF antigen assay and the functionality of vWF with a glycoprotein (GP) Ib binding assay, a collagen-binding assay, or a ristocetin cofactor activity (RiCof) or ristocetin-induced platelet agglutination (RIPA) assays (see above). Factor VIII levels are also performed because factor VIII is bound to vWF. Deficiency of vWF can, therefore, lead to a reduction in factor VIII levels, which explains the elevation in PTT time.
· Normal levels do not exclude all forms of vWD, particularly type 2, which may only be revealed by investigating platelet interaction with subendothelium under flow (PAF). A platelet aggregation assay will show an abnormal response to ristocetin with normal responses to the other agonists used. A platelet function assay (PFA) will give an abnormal collagen/ adrenaline closure time and, in most cases (but not all), a normal collagen/ ADP time. Type 2N can only be diagnosed by performing a "factor VIII binding" assay.
· Note: Detection of vWD is complicated by vWF being an acute phase reactant, with levels increasing in infection, pregnancy, and stress.
· Treatment: for patients with vWD type 1 and vWD type 2A, desmopressin (DDAVP) is recommended for use in cases of minor trauma or in preparation for dental or minor surgical procedures. DDAVP stimulates the release of von Willebrand factor (vWF) from the Weibel Palade bodies of endothelial cells, thereby increasing the levels of vWF (as well as coagulant factor VIII) 3 to 5-fold. DDAVP is available as an intranasal or intravenous form of administration.
· DDAVP is contraindicated in vWD type 2b because of the risk of aggravated thrombocytopenia and thrombotic complications. DDAVP is probably not useful in vWD type 2M and is rarely helpful in vWD type 2N. It is totally ineffective in vWD type 3.
· Haemophilia A: the most common hereditary coagulation disorder; deficiency or absence of factor VIII.
· Occurs in about 1:10,000 people.
· X-linked recessive; point mutations or deletion of factor VIII gene.
· It manifests in males; female carriers are asymptomatic.
· Clinical features: depends on severity; presentation in early childhood; family history often positive (manifests in men; X-linked recessive); recurrent deep-seated intramuscular hematomas and hemarthrosis (especially knees, ankles, and elbows); excessive bleeding after a trivial trauma; spontaneous and excessive post-traumatic hemorrhage; epistaxis; hematuria; bleeding in the retroperitoneum and the brain can be life-threatening.
· Severity (normal level of factor VIII is 50 – 150%):
· a) Factor VIII level < 1% (severe; 70% of patients): frequent and spontaneous deep tissue hemorrhages and hemarthrosis.
· b) Factor VIII level 1 – 5% (moderate; 15% of patients): excessive hemorrhage after mild to moderate injury; occasional hemarthrosis; spontaneous bleeding infrequent.
· c) Factor VIII level >5% and <50% (mild; 15% of patients): excessive hemorrhage only after significant trauma or surgery.
· Lab features Prolonged APTT.
· Diagnosis: with factor VIII (FVIII) assay.
· Treatment: Most severe hemophilia patients require regular supplementation with IV recombinant or plasma concentrate Factor VIII. The prophylactic treatment regime is highly variable and individually determined. Apart from ‘routine’ supplementation, extra factor concentrate is given around surgical procedures and after trauma.
· Haemophilia B: Hemophilia B (also called Christmas disease) is a blood clotting disorder caused by a mutation of the factor IX gene, leading to a deficiency of factor IX. It is the second-most common form of hemophilia and rarer than hemophilia A. The factor IX gene is located on the X – chromosome. Hemophilia B is an X–linked recessive trait. Thus, as in hemophilia A, usually, only males are affected. It affects one in 20,000–30,000 men.
· Factor IX deficiency leads to an increased tendency for hemorrhage, such as in response to mild trauma or even spontaneously, such as in joints (hemarthrosis) or muscles.
· Treatment (bleeding prophylaxis) is by intravenous infusion of factor IX. Factor IX has a longer half-life than factor VIII (deficient in hemophilia A), so factor IX can be transfused less frequently.
· Acquired Haemophilia: acquired hemophilia is a rare but potentially life-threatening bleeding disorder caused by the development of autoantibodies (inhibitors) directed against plasma coagulation factors, most frequently factor VIII (FVIII).
· Signs & symptoms: hemorrhages into the skin, muscles, or soft tissues and mucous membranes. However, intra-articular (joint) bleeding episodes are uncommon.
· Lab tests: on patients not taking heparin, the bleeding time, prothrombin time (PT), and platelet count are normal, but the activated partial thromboplastin time (aPTT) typically shows a prolongation that is not reversed on a correction study. Reduced FVIII levels and evidence of an FVIII inhibitor are critical to the diagnosis of acquired hemophilia A. The acquired inhibitor should be quantified to project the severity of the disorder and the risk of hemorrhagic complications. Other factor levels should be determined to establish inhibitor specificity. Lupus anticoagulant (e.g., dilute Russell viper venom time and the kaolin clotting time) should be checked if aPTT values during the mixing study are similar at time 0 and after incubation at 37°C.
· Treatment: treating the underlying disorder or discontinuing an offending drug. High-dose infusions of FVIII may help hemostasis in patients with low-titer inhibitors. Patients with very low inhibitor titers (< 3 Bethesda units [BU]) and residual FVIII activity may also benefit from treatment with desmopressin (synthetic vasopressin (antidiuretic hormone)). Moderate to severe bleeding in patients with inhibitor titers of less than 5 BU can be treated with recombinant FVIII porcine sequence (Obizur), which is approved for as it is less likely than human FVIII to be neutralized by the inhibitors that occur in acquired hemophilia A. Patients with severe bleeding and inhibitor titers of 5 BU or higher should receive therapy with either recombinant factor VIIa or an activated prothrombin complex concentrate (APCC). Eradication of the inhibitor with immunosuppression should be initiated as soon as the diagnosis of acquired hemophilia is established with methylprednisolone or prednisone. Adding oral cyclophosphamide (50-150 mg/d) can increase the response rate. Rituximab may be considered in cases of resistance to or intolerance of standard immunosuppressive therapy. Cyclosporine is particularly useful in patients with underlying systemic lupus erythematosus (SLE).
No comments:
Post a Comment